



Presentamos un caso de leishmaniasis visceral con un síndrome hemofagocítico asociado en un niño trasplantado renal, cuyo diagnóstico fue difícil dada la coincidencia con elevación marcada de la proteína C reactiva-virus de Epstein-Barr. A pesar de su sintomatología, su estado general era bueno y estable, por lo que se decidió no tratar su síndrome hemofagocítico e iniciar tratamiento con anfotericina B liposomal intravenosa. Ante la no respuesta a dicho tratamiento se asoció miltefosina, con mejoría de la sintomatología hasta su curación, sin efectos secundarios ni afectación de la función del injerto renal.
Se pretende llamar la atención sobre este cuadro infeccioso cada vez más frecuente y que se extiende a distintos países por múltiples factores. Además, tiene la particularidad de ser un paciente pediátrico trasplantado renal con mala respuesta al tratamiento inicial, lo que nos supuso muchas dudas dado que este no tiene un protocolo único establecido, ni en la medicación utilizada, ni en la dosificación ni en el tiempo de tratamiento.
INTRODUCCIÓN
La leishmaniasis visceral (LV) es una zoonosis endémica en los 5 continentes. Está causada por protozoos del género Leishmania, y se transmite por la picadura de flebotomos hembras infectados.
Se asocia a situaciones de pobreza, con alto índice de malnutrición y debilidad del sistema inmunitario, así como a factores ambientales como la deforestación, la construcción de presas, los sistemas de riego y la urbanización. La Organización Mundial de la Salud1 estima que cada año se producen entre 700.000 y 1 millón de nuevos casos y entre 20.000 y 30.000 defunciones, a pesar de que solo una pequeña parte de las personas infectadas desarrolla la enfermedad.
Actualmente hay un aumento en todo el mundo de condiciones que favorecen su desarrollo, entre otras, la gran migración y el desplazamiento de personas no inmunizadas, la deforestación y el cambio climático. Esto, a pesar de los programas de eliminación de la enfermedad en los países con mayor número de casos, probablemente influye en su incremento en zonas no tan habituales y en una población inmunodeprimida, como son los receptores de un trasplante de órgano sólido.
El tratamiento óptimo en estos pacientes aún no está definido, y menos aún en pacientes pediátricos.
Presentamos un caso de LV en un niño trasplantado renal, que planteó serias dudas en el diagnóstico diferencial con un síndrome asociado poco frecuente, la linfohistiocitosis hemofagocítica (HLH), cuyo tratamiento es muy diferente. Tratado inicialmente con anfotericina B liposomal, medicación que sustituyó a los antimoniales tras su aprobación por la Food and Drug Administration en 1997 para el tratamiento de la LV2, asociamos miltefosina a dicho tratamiento, con buena evolución a pesar de una recaída posterior.
CASO CLÍNICO
Niño de 6 años de edad, trasplantado renal de donante vivo emparentado 2,5 años antes, tras 4 meses en hemodiálisis por estar afectado de insuficiencia renal terminal secundaria a tubulopatía (hipomagnesemia con hipercalciuria y nefrocalcinosis familiar por mutación del gen de la claudina 19).
Realizaba tratamiento inmunosupresor con tacrolimus, micofenolato mofetilo (MMF) y prednisona a dosis mínimas (5 mg en días alternos), tras tratamiento de inducción con 2 dosis de basiliximab.
La evolución postrasplante había sido muy buena, con función renal normal del injerto. Destacaba:
• Un ingreso a los 4 meses del trasplante por cuadro neumónico adquirido en la comunidad, con resolución total tras antibioterapia.
• Positivización a los 6 meses del trasplante, con leve sintomatología digestiva, de PCR-CMV y virus BK en plasma y orina, que se negativizaron tras 3 semanas de tratamiento con valganciclovir oral y disminución transitoria de la inmunosupresión.
Ingresó en nuestro centro procedente de otro hospital, por fiebre de 9 días de evolución (hasta 40 °C) y pancitopenia, motivo por el que le habían disminuido la dosis de MMF.
A la exploración tenía un buen estado general, aunque destacaba una notable palidez mucocutánea, sin adenopatías, exantemas ni otras lesiones.
La auscultación pulmonar era normal, con corazón rítmico, sin soplos y taquicardia de 110 lpm, aproximadamente. El abdomen era blando y depresible, sin masas ni visceromegalias al inicio, comenzando a palparse esplenomegalia a los 4-5 días del ingreso, con un máximo de 4 cm del reborde costal.
Analíticamente destacaba:
• Pancitopenia: hemoglobina (Hb), 5,8 g/dl; leucocitos, 1.890/μl (44% PMN), 57.000 plaquetas/μl. Sin presencia de blastos en sangre periférica.
• Función renal con ligero aumento de la creatinina sobre sus valores previos: de 0,6 a 0,87 mg/dl, con niveles plasmáticos de tacrolimus en rango terapéutico habitual.
• Proteína C reactiva (PCR) de 80,6 mg/l.
• Enzimología hepática y colesterol, normales; triglicéridos, 394 mg/dl; anticuerpos antinucleares y anticitoplasmáticos, negativos.
• Estudio de coagulación, normal; fibrinógeno, 160 mg/dl.
• Metabolismo del hierro: sideremia, 18,4 μg/dl; IST, 6%; transferrina, 229 mg/dl; ferritina, 2.356 μg/l (sin haber recibido transfusiones).
• PCR citomegalovirus (CMV), negativa; PCR virus Epstein- Barr (VEB), 162.192 copias/ml.
• Hemocultivos y urocultivos, negativos.
• Virus influenza A y B en aspirado nasofaríngeo, negativos.
• Radiografía anteroposterior de tórax, sin alteraciones.
• Ecografía abdominal: injerto renal sin anormalidades; esplenomegalia homogénea.
• Tomografía con contraste intravenoso (i.v.) de abdomen, cuello y tórax, sin adenopatías con significación radiológica. Injerto renal en fosa ilíaca derecha, con adecuada diferenciación corticomedular, sin lesiones focales, con pelvis ligeramente ectásica. Hepatoesplenomegalia, con probable edema periportal adyacente a la bifurcación portal.
• Estudio de punción-aspirado de médula ósea, pancitopenia. Celularidad medular conservada con moderada dishemopoyesis inespecífica y discretos signos de reactividad. Algunas imágenes de hemofagocitosis.
• Microbiología/serología de virus: negativa para Mycoplasma pneumoniae y Chlamydia pneumoniae,Coxiella burnetti, herpes virus 6, parvovirus y virus de la inmunodeficiencia humana. IgG positiva para CMV y VEB, y positiva a Leishmania en un título de 1/128, con IgM negativa.
Evolución y tratamiento
En los primeros días de ingreso se diagnostica de infección por VEB, dado el alto valor de la PCR, disminuyéndose aún más la inmunosupresión (se suspende el MMF y se disminuye la dosis de tacrolimus para niveles plasmáticos en torno a 3-4 ng/ml). Tras una dosis de rituximab (375 mg/m2 i.v.) desciende rápidamente dicha PCR, así como las inmunoglobulinas del paciente, y desaparecen los linfocitos B-CD19+.
A pesar de ello, la sintomatología del niño persiste, con picos de fiebre de 40° axilar, difícil de controlar con antitérmicos, escalofríos, anemia y plaquetopenia marcadas (requirió 2 transfusiones de concentrado de hematíes y 4 de plaquetas), y, en ocasiones, aparición de máculas hiperémicas (fig. 1) relacionadas con el pico febril, que desaparecían al descender la temperatura. En un nuevo estudio de médula ósea se positiviza la PCR a leishmanias, y también lo hizo en sangre; BAAR (bacilos ácido alcohol resistentes), negativos.
Figura 1. Lesiones maculares que aparecieron en la evolución.
Se constata un aumento de ferritina de hasta 5.597 μg/l (ya tras las transfusiones).
Al estudiar inmunodeficiencias destacan, además de leucopenia, anemia y trombocitopenia: niveles elevados de CD25 soluble (12,74 ng/ml; valores normales 0-7,5), valores elevados de C1q y C5 (normales los C3 y C4) y niveles disminuidos de IgA, con IgG e IgM normales. En el estudio de subpoblaciones linfocitarias hay ausencia de células B CD19 (secundario a tratamiento con rituximab), con escaso número de células natural killer (NK) (< 1%), número normal de células T (CD4 y CD8) y expresión de perforinas positiva.
Diagnosticado de LV, sin poder descartar un síndrome hemofagocítico asociado, se inició tratamiento con anfotericina B liposomal3,4 i.v. a 3 mg/kg/día y 1 dosis de 500 mg/kg de inmunoglobulinas inespecíficas.
Tras 10 dosis de anfotericina y 1 dosis acumulativa de 30 mg/kg, no se apreció mejoría clínica ni analítica, persistiendo PCR positiva a leishmanias en plasma y médula ósea. Se asoció al tratamiento paromomicina oral, a la espera de conseguir miltefosina oral, dada la no disponibilidad de paromomicina parenteral en nuestro hospital, y se espació la anfotericina B a dosis semanales5-7. A la semana, sin ninguna mejoría con la paromomicina, se inició tratamiento con miltefosina (2 mg/kg/día), que recibió durante 4 semanas, que se asoció a la anfotericina, ahora semanal, hasta completar una dosis acumulativa total de 50 mg/kg (7 dosis semanales, 17 dosis en total).
La fiebre desapareció al mes del inicio del tratamiento con anfotericina i.v. y a los 15 días del inicio de la miltefosina, y la primera PCR negativa se obtuvo 10 días después (fig. 2).
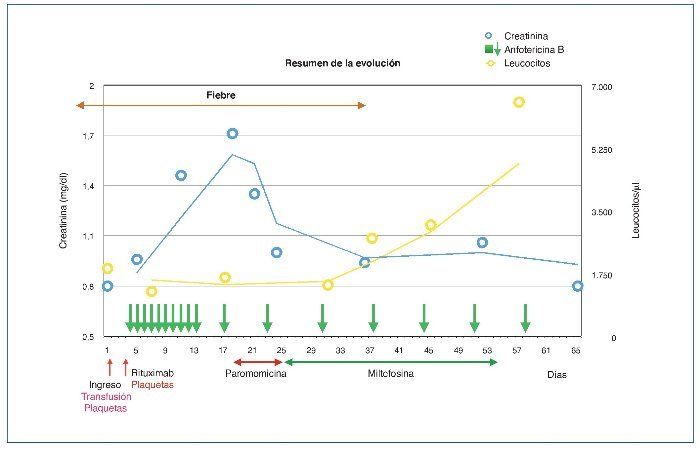
Figura 2. Resumen de la evolución y tratamiento.
La función del injerto permaneció estable, con ligero ascenso de la creatinina plasmática durante el cuadro clínico y tras las dosis de anfotericina B, que al finalizar y suspender dicho tratamiento fue de 0,8 mg/dl. No se dejó la profilaxis, para evitar posibles recaídas de leishmaniasis.
El niño reingresó 1,5 meses después por nueva positivización de la PCR a leishmanias en control ambulatorio, coincidiendo con moderada leucopenia (3.670 leucocitos/μl), ascenso de la velocidad de sedimentación globular y anorexia.
Se reinició tratamiento con anfotericina B liposomal a dosis algo más elevada, 4 mg/kg/día, durante 5 días consecutivos, seguidas de 2 dosis semanales, y se negativizó la PCR en sangre periférica a la semana de tratamiento, que ha persistido negativa hasta la actualidad.
A las 6 semanas se detectó nuevamente anemia grave con descenso de la Hb hasta 5,5 g/dl, que requirió varias transfusiones de concentrado de hematíes y tratamiento con eritropoyetina.
En este caso, las PCR a CMV, VEB, BK y leishmania eran negativas, y se comprobó una positivización del parvovirus B19, que desaparició tras la disminución temporal de la inmunosupresión.
Actualmente, tras 2 años de la finalización del segundo ciclo de tratamiento, el paciente sigue libre de infección, asintomático y con injerto normofuncionante y estable.
DISCUSIÓN
Aunque nuestro centro atiende a una zona teóricamente endémica de leishmaniasis, la cuenca del Mediterráneo, desde que se inició el programa de trasplante, tras 309 trasplantes realizados, es el primer caso de LV en uno de estos pacientes.
En la bibliografía se encuentran trabajos en los que se recogen la asociación LV/HLH, pero son escasos en pacientes pediátricos8 y no se encuentra en niños inmunosuprimidos como el que presentamos.
Su cuadro clínico, fundamentalmente fiebre elevada, con pancitopenia y esplenomegalia, está descrito entre los síntomas habituales de la LV, aunque son síntomas, por otro lado, que pueden estar presentes en otras infecciones oportunistas en pacientes inmunocomprometidos o —como fue nuestro caso— pueden hacer sospechar un posible cuadro linfoproliferativo al unirse con una positividad elevada de la PCR al VEB.
El síndrome hemofagocítico reactivo, o HLH secundaria8, es un síndrome que se caracteriza por una activación anormal del sistema inmunitario después de una estimulación excesiva de las células NK y los linfocitos T CD8, que produce daño celular y proliferación y activación de macrófagos benignos con hemofagocitosis en el sistema reticuloendotelial. Se inicia, típicamente, por una infección, tanto en individuos predispuestos genéticamente como en casos esporádicos, y las infecciones más frecuentes que lo desencadenan son las virales y, dentro de ellas, la causada por el VEB, aunque también se puede asociar a CMV, parvovirus, virus herpes, etc. A veces, aunque menos frecuentemente, el factor desencadenante pueden ser infecciones bacterianas (Brucella, tuberculosis, etc.) o parasitarias (entre ellas, la leishmaniasis). Fundamentalmente, es un síndrome pediátrico, aunque puede encontrarse a cualquier edad. Al inicio se presenta como una enfermedad febril, que puede afectar a distintos órganos mimificando un proceso infeccioso. Su diagnóstico debe cumplir, en un contexto clínico adecuado, al menos 5 de estos 8 criterios3,4: 1) fiebre ≥ 38,5 °C; 2) esplenomegalia; 3) citopenias, con al menos 2 de lo siguientes valores: Hb < 9 g/dl, plaquetas < 100.000/μl, recuento absoluto de neutrófilos < 1.000/μl; 4) ferritina ≥ 500 ng/dl; 5) triglicéridos en ayunas > 265 mg/dl y/o fibrinógeno < 150 mg/dl; 6) hemofagocitos en médula ósea, bazo, ganglios linfáticos o hígado sin neoplasia maligna; 7) disminución o ausencia de actividad de células NK, y 8) aumento del CD25 soluble 2 desviaciones estándar por encima de los límites del laboratorio ajustados para la edad. No obstante, dada la alta mortalidad de este síndrome en ausencia de un tratamiento apropiado, no siempre se requieren todos ellos para iniciarlo. Nuestro paciente cumplía 6 de estos criterios.
Inicialmente se consideró su posible asociación al VEB pero, al no mejorar el cuadro con la desaparición de dicho virus y obtener la positividad a leishmanias, se siguió considerando dicho diagnóstico secundario, en este caso, al parásito.
El tratamiento de la HLH primaria es la quimioterapia; el protocolo más utilizado incluye ciclosporina, etopósido y dexametasona5. En la forma secundaria, si el paciente no está gravemente enfermo (no tiene deterioro cardiovascular, pulmonar, renal, hepático ni neurológico), el tratamiento se fundamenta en la identificación y el tratamiento de la enfermedad subyacente, y puede ser posible evitar la quimioterapia, aunque esto es la excepción más que la regla4,6.
En nuestro paciente, su situación clínica estable y con un aceptable estado general, a pesar de sus datos clínicos descritos, nos decidió a no iniciar tratamiento específico del síndrome hemofagocítico y tratar solo la leishmania.
Se inició el tratamiento, como recomiendan otros artículos y guías7,9-11, con anfotericina B liposomal diaria. Estos indican una dosis acumulativa total de 40 mg/kg. En nuestro caso, tras alcanzar una dosis de 30 mg/kg y continuar el niño con la misma sintomatología, sin apreciar mejoría clínica ni analítica, se decidió asociar miltefosina, con una buena respuesta a los 15 días del inicio del tratamiento combinado. Hay escasos trabajos en la bibliografía12 de la utilización conjunta de ambos tratamientos en adultos, alguno con el resultado final de fallecimiento del paciente, precisamente por un cuadro de HLH13, pero no hemos encontrado ninguno en niños. En nuestro paciente pediátrico, la evolución fue muy buena, la enfermedad desapareció y se mantuvo una buena función del injerto tras un moderado ascenso temporal de la creatinina durante el tratamiento.
Como se ha descrito en otros trabajos, las recaídas son frecuentes, como sucedió en nuestro caso, pero no hay acuerdo en el uso ni indicaciones de profilaxis7. La respuesta en nuestro paciente a un nuevo ciclo, esta vez corto y sin miltefosina, de anfotericina B liposomal fue excelente.
No hubo ningún tipo de mejoría al asociar al tratamiento la paromomicina oral, lo que consideramos que se debe a la no absorción de dicho medicamento por esa vía de administración, por lo que, de esta manera, no resulta útil para este tipo de infección.
Esperamos que nuestra experiencia pueda ayudar en el tratamiento de otros pacientes que, como en este caso, tardan en responder. No hemos encontrado en la bibliografía un tiempo estimado de respuesta, qué hacer si cumple criterios de síndrome hemofagocítico y qué perfil de seguridad se puede esperar con un tratamiento combinado de anfotericina B liposomalmiltefosina.
Conflicto de intereses
Los autores declaran que no tienen conflicto de intereses potencial relacionado con los contenidos de este articulo.
Correspondencia: Julia Fijo
Unidad de Nefrología Pediátrica.
Hospital Infantil. HH. UU. Virgen del Rocío.
Avenida Manuel Siurot, s/n. 41013 Sevilla.
mj.fijo.sspa@juntadeandalucia.es
Revisión por expertos bajo la responsabilidad de la Sociedad Española de Nefrología.


