

La epigenética estudia los mecanismos que regulan la expresión génica sin modificar la secuencia del ADN. Estos mecanismos, que incluyen procesos moleculares como la metilación de ADN, modificaciones de las histonas y los ARN no codificantes como los micro-ARN, permiten la activación o la represión de los genes jugando un papel relevante en los numerosos procesos biológicos. Diferentes alteraciones de los patrones epigenéticos tienen importantes consecuencias fisiológicas y son un componente central en el desarrollo de muchas enfermedades humanas. La epigenética en nefrología está aún en fases muy iniciales de desarrollo. La presente revisión pretende ser un acercamiento a esta disciplina y describir los recientes hallazgos de la epigenética relacionados con la enfermedad renal crónica, la nefropatía diabética y el desarrollo de la proteinuria. Se describen las diferentes modificaciones epigenéticas en estas entidades y las posibilidades que ello supone en el desarrollo, diagnóstico y potenciales tratamientos.
INTRODUCCIÓN
Se estima que en el humano existen unos 200 tipos de células diferentes. Todas ellas con el mismo genoma y, por tanto, con la misma cadena de ADN. Conceptualmente, la epigenética estudia cómo se expresa este ADN. Tan solo en los últimos años se están empezando a conocer los fundamentos de esta diversidad y plasticidad y comenzando a intuir la gran complejidad de los diferentes mecanismos reguladores implicados. Existe un control por encima de la secuencia del ADN, una serie de modificaciones que, sin cambiar la secuencia del ADN, decidirán qué genes se expresarán sellando el destino celular. No solo la epigenética tiene un papel primordial en la identidad celular, mediante mecanismos epigenéticos, también la expresión génica de las células puede ser modulada por factores externos (ambiente). Investigaciones en este campo han evidenciado una estrecha relación entre modificaciones epigenéticas y el desarrollo de enfermedades, y es el motivo de que la investigación básica haya comenzado a trasladarse a la investigación clínica. Hoy se sabe que estos cambios epigenéticos son también susceptibles de ser modificados, lo que abre un amplio horizonte de posibilidades terapéuticas hoy en día insospechadas.
No cabe duda de que en los próximos años la epigenética será la gran protagonista, tanto en la investigación básica como en la clínica. La presente revisión pretende ser un acercamiento al clínico de esta fascinante ciencia, haciendo especial referencia a la relación entre epigenética y nefrología. Por cuestión de espacio, se ha centrado fundamentalmente en el campo de la enfermedad renal crónica (ERC), la nefropatía diabética (ND) y el desarrollo de la proteinuria, bien es cierto que la epigenética también es centro de interés en otros campos como el fracaso renal agudo, la inmunología, el trasplante renal, etc.
UNA BREVE HISTORIA DE LA EPIGENÉTICA
Tanto Waddington, acuñando el término epigenética en 1942, como Nanney, 16 años más tarde, aportando el concepto de la estabilidad de los estados de expresión a lo largo de la división celular, son los pioneros en este campo. En su inicio, la epigenética se definió como el estudio de las interacciones causales entre genes y sus productos que llevan al fenotipo durante el desarrollo1. Aún en los años sesenta del pasado siglo, había opiniones divergentes de cómo se producía la especificidad celular entre las más de 200 células somáticas existentes, partiendo de una sola célula fertilizada; por ejemplo, ¿una célula muscular podría reconfigurar sus programas de expresión génica para desarrollar la actividad neural o para la formación de hueso?2.
Laskey y Gurdon en 1970 despejaron la duda demostrando cómo el núcleo de una célula somática era competente en dirigir la embriogénesis cuando era introducida en el óvulo enucleado. Quedaba claro que el repertorio de expresión génica visto en las células somáticas no era el resultado de delecciones o mutaciones en la secuencia del ADN germinal cuando era transmitido a las células somáticas, el ADN era el mismo3. Estudios posteriores mostraron cómo cambios en la metilación del ADN modificaban la expresión génica sin alterar la secuencia de nucleótidos y además estos patrones podían ser mantenidos en sucesivas divisiones celulares. En la actualidad, la epigenética se define más exactamente como “el estudio de las modificaciones genéticas estables que resultan en cambios en la expresión y función génica sin una alteración correspondiente en la secuencia de ADN”1,4,5. Además de la metilación en el ADN, hoy en día se reconocen como mecanismos epigenéticos, las modificaciones en las proteínas que están estrechamente asociadas con el ADN, las histonas, y los micro-ARN (mi-ARN).
La adquisición progresiva del conocimiento del mecanismo molecular que gobierna la identidad celular y el hecho de que en parte pueda ser modulado y revertido, el que diversos organismos tengan mecanismos moleculares comunes que contribuyen al control epigenético de la expresión del gen, una mayor comunicación entre diferentes ramas de la ciencia y la mayor disposición y generalización de la tecnología necesaria, entre otros, han contribuido a una enorme expansión de esta disciplina siendo de interés en numerosos y aparentemente dispares campos de la ciencia. Si bien, a lo largo de 1995 se referenciaron 114 entradas en PubMed con el término epigenetic, a lo largo de 2016 se referenciaron 7.851 nuevas entradas.
Aunque las modificaciones epigenéticas son estables, pueden ser moduladas por numerosas condiciones fisiológicas, patológicas y ambientales (revisadas en las referencias 5 y 6). Se ha demostrado cómo gemelos monocigotos, idénticos en la secuencia del ADN e indistinguibles epigenéticamente en los primeros años, a lo largo de la vida presentarán significativas diferencias a nivel de las marcas epigenéticas7. Por otra parte, otros estudios han comprobado cómo el patrón de metilación del ADN y las marcas de histonas como H3K4me1, difieren entre la edad temprana de la vida y la edad longeva8,9. Todo ello, en conjunto, supone que más que genéticamente predeterminada, nuestra vida está epigenéticamente determinada. La dieta y otras influencias ambientales pueden influir en nuestra vida modificando la información epigenética y la de nuestra descendencia. Estos nuevos hallazgos proporcionan una mejor comprensión de los mecanismos involucrados en el inicio y desarrollo de la enfermedad o del envejecimiento y, dada su naturaleza reversible, abren nuevos caminos para la intervención terapéutica.
MAQUINARIA EPIGENÉTICA
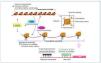
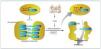
En la célula eucariota, el ADN está empaquetado en el núcleo de forma altamente organizada. La unidad básica de cromatina la compone un octámero, el nucleosoma, formado por 2 copias de cada una de las 4 histonas que lo componen, H2A, H2B, H3 y H4, alrededor del cual está el ADN enrollado (145-147 pares de bases [pb]). Este complejo de nucleoproteínas, altamente conservado, se produce esencialmente cada 200 pb a lo largo de todo el genoma y estas estructuras pueden estar más o menos compactadas. Dependiendo del grado de compactación de la cromatina, el grado de transcripción varía, a menor grado de compactación esta será más accesible y transcripcionalmente más activa (eucromatina) que cuando adopta un estado más compacto (heterocromatina)10 (fig. 1).
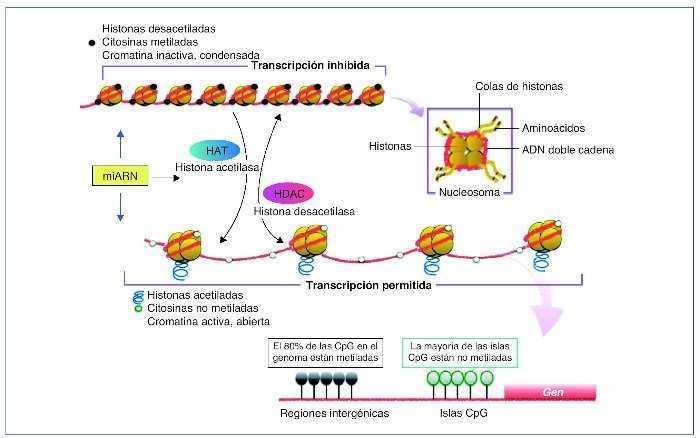
Figura 1. Representación de las diversas modificaciones epigenéticas.Sin modificar la secuencia del ADN, la expresión génica de una determinada célula variará. Estas modificaciones darán identidad a la célula. Si bien son estables, pueden ser modificadas mediante estímulos externos. Los patrones de metilación se pueden heredar y transmitir a futuras generaciones. Tanto la metilación de ADN como la modificación de histonas y los mi-ARN están sometidas a una estrecha autorregulación. En el CpG dinucleótido citosina-guanina es donde se lleva a cabo la metilación del ADN. Las islas CpG se suelen situar en la zona promotora del gen. Un alto grado de metilación se asocia con el silenciamiento de genes. CpG: citosina-fosfato-guanina.
Metilación del ADN
La metilación del ADN es la modificación epigenética más conocida. Consiste en la adición de un grupo metilo (-CH3) a la citosina en 5-(meC), ocurre en las citosinas seguidas de guaninas (CpG [citosina-fosfato-guanina] dinucleótidos) y usualmente se asocia con silenciamiento genético11,12. En el genoma humano, los dinucleótidos CpG habitualmente se concentran en regiones llamadas islas CpG que, preferentemente, se encuentran en las regiones promotoras del gen, estando generalmente no metiladas. Las CpG localizadas fuera de las islas, por lo general, están metiladas y se cree que participan en la estabilidad del cromosoma. Además se ha evidenciado CpG metilado a nivel intragénico, sugiriéndose un papel regulatorio del splicing13. La metilación de la CpG se lleva a cabo mediante las enzimas metiltransferasas (DNMT), siendo las más conocidas DNMT3A, DNMT3B y DNMT1. Las DNMT3A y 3B estarían implicadas en la metilación de novo y la DNMT1, por su apetencia por el ADN hemimetilado, aseguraría el patrón de metilación en la replicación a través de la división celular. La DNMT1 es la más abundante en las células somáticas y sería la responsable del mantenimiento de la metilación del ADN13,14.
La represión de la transcripción, mediada por la metilación del ADN, puede ser producida de 2 formas no excluyentes entre sí. La primera, por interferencia directa del grupo metilo para la unión de una proteína específica a esa secuencia, por ejemplo, la de un factor de transcripción. La segunda, por la unión de las proteínas MBD (methyl-CpG-binding domain) que son atraídas por el grupo metilo CpG. Estas proteínas (de las que se han descrito 4 tipos) actuarían en conjunto mediante el reclutamiento de enzimas modificadoras de histonas, como las histonas desacetilasas (HDAC1 y HDAC2), las metiltransferasas y otros complejos remodeladores de la cromatina, provocando una mayor compactación de la cromatina (heterocromatina) y, por tanto, mantendrían y reforzarían el estado represor de la expresión génica11,14,15. Recientemente se ha demostrado cómo ciertos mi-ARN (v. más adelante) podrían ejercer un papel regulador-modulador de este silenciamiento génico, al menos por 2 mecanismos, bien interactuando con las metiltransferasas, contribuyendo a la metilación del ADN; bien mediante la destrucción de cualquier potencial ARNm (ARN mensajero) que hubiera podido eludir la represión16. Los mecanismos responsables de la desmetilación activa, es decir, en ausencia de división celular, en mamíferos aún no han sido completamente dilucidados. Uno de los mejor documentados es el de la oxidación de la 5-meC por parte de enzimas de la familia TET (ten-eleven-translocation), compuesta por 3 miembros, TET1, TET2 y Tet317.
Modificación de las histonas
Dentro de un nucleosoma, cada una de las histonas, altamente conservadas, contiene un dominio globular estructurado, que interactúa con las otras histonas y el ADN. Las histonas poseen unas colas flexibles que sobresalen de la superficie lateral del nucleosoma. Las colas flexibles son muy básicas y son los sustratos para numerosas modificaciones. Dado que hay al menos 4 residuos de aminoácidos que están sujetos a modificación, lisina (K), serina (S), treonina (T) y arginina (R), y más de 6 tipos de modificaciones: metilación, acetilación, fosforilación, ubiquitinación, biotinilación, sumoilación e isomerización de la prolina, el número de posibles combinaciones es extraordinariamente elevado18,19. Debido a que las distintas modificaciones de las histonas se correlacionan con estados transcripcionales específicos, se ha desarrollado la hipótesis de código histona. Esta hipótesis sugiere que los patrones específicos de modificaciones se leerían como un código de barras molecular para reclutar la maquinaria celular que produce un estado distinto de la cromatina20. Por ejemplo, los residuos de histona pueden modificarse de manera que promuevan la transcripción o el silenciamiento, como en el caso de H3K9, que puede ser trimetilado (para promover la represión génica) o acetilado (para promover la transcripción activa)19 (fig. 1).
Las modificaciones más ampliamente estudiadas a nivel de las histonas H3 y H4 incluyen la acetilación y desacetilación, mediadas por las histonas acetilasas (HAT) y desacetilasas (HDAC), y la metilación, mediada por las histonas metiltransferasas (HMT) y sus correspondientes histonas demetilasas (HKDM)19.
Micro-ARN
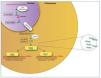
Los mi-ARN son pequeños transcritos no codificantes (de 19 a 25 nucleótidos de longitud), cuya principal función es la de regular la expresión génica uniéndose a los ARNm diana, dirigiéndolos a la degradación o evitando su traducción16, aunque también se ha sugerido un rol en el control de la transcripción y el splicing21. Los mi-ARN están evolutivamente conservados entre las diferentes especies y son cruciales en importantes funciones celulares como el desarrollo, la proliferación y diferenciación, la apoptosis y la respuesta al estrés22. Se han descrito más de 1.000 mi-ARN, que se codifican en las regiones intergénicas o en los exones o intrones de genes codificantes de proteínas. El mi-ARN se sintetiza como un largo transcrito en el núcleo por la ARN polimerasa. Posteriormente es escindido por una ARNsa formando un precursor premi-ARN, que es transportado al citoplasma por una exportina para posteriormente ser procesado por una segunda ARNsa (Dicer). Dicer escinde el premi-ARN produciendo moléculas de 22 bp de doble cadena de mi-ARN. Una de las cadenas se unirá a ARN-inductor de silenciamiento o RISC (RNA-induced silencing complex) mientras que la cadena inactiva será degradada. El grado de complementariedad entre los mi-ARN y el ARNm será determinante como mecanismo regulador. Una perfecta complementariedad con el ARNm provocará la fragmentación del ARNm, por el contrario, una unión imperfecta llevará a la represión translacional del ARNm, que posteriormente podrá ser degradado o bien traducido22.
Los mi-ARN también forman parte de la maquinaria epigenética interviniendo en la regulación de la metilación del ADN y de la acetilación de las histonas estableciendo un fino control y feedback regulador de la expresión génica22,23 (fig. 2).
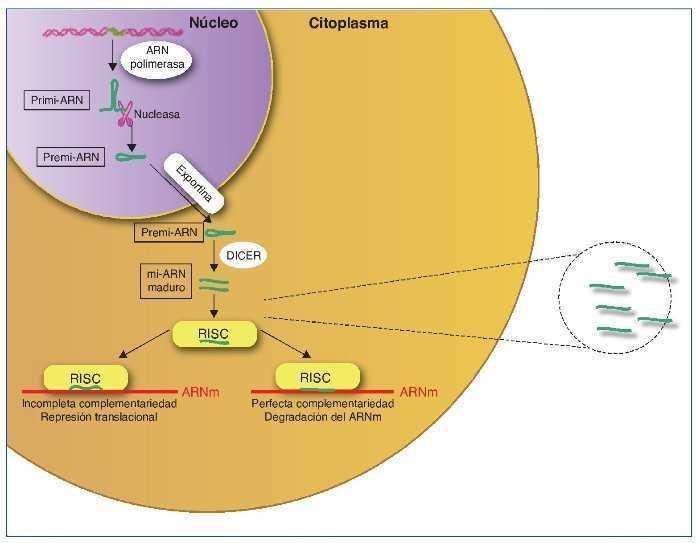
Figura 2. Síntesis de los mi-ARN. Los mi-ARN pueden salir de la célula al medio extracelular y ser hallados en los diferentes líquidos biológicos, y son muy duraderos y resistentes a la degradación; su uso como biomarcadores es objeto de intensos estudios.
EPIGENÉTICA EN NEFROLOGÍA
Desde hace muy pocos años, la epigenética es objeto de estudio en la patología renal. Hasta el momento, los mayores avances se han producido en la ERC y en la ND, entre otros.
Epigenética y enfermedad renal crónica
Estudios de metilación global de ADN realizados en sangre periférica en pacientes con ERC y con diagnóstico de arteriosclerosis han mostrado resultados contradictorios, tanto hipermetilación como hipometilación de ADN. Sin embargo, cuando se ha investigado el tejido afectado en muestras extraídas de arterias coronarias tras realizar revascularización, se ha apreciado una importante hipometilación de las islas CpG24. Estos resultados ponen de manifiesto la complejidad del abordaje de este tipo de estudios. Los diferentes resultados se podrían explicar por diferentes motivos, entre ellos diferentes técnicas de laboratorio, ausencia de estandarización, diferente metodología y diferencias en la definición de arteriosclerosis. Además, está en debate la validez del estudio de metilación en sangre periférica, ya que el patrón de metilación diferiría respecto al que se produce a nivel de la lesión, al tratarse de diferentes células24.
Independientemente de la causa inicial, la progresión de la ERC conduce a la glomeruloesclerosis y a la fibrosis intersticial, que se caracteriza por un extenso tejido cicatricial que abocará a la ERC terminal. Este proceso estaría en gran parte mediado por fibroblastos que originariamente procederían de la estirpe epitelial original y que por un proceso de transición, influenciado por factores locales como factores de crecimiento (TGF, FGF-2, angiotensina II), interleucinas, citocinas, etc., alcanzarían un estatus mesenquimal25,26. Estas células de fenotipo diferente al inicial y por tanto con propiedades biológicas diferentes conducirían a la fibrosis progresiva y a la ERC terminal. Este proceso de transición conocido como EMT (epitelial mesenchymal transition) estaría regulado por mecanismos epigenéticos26,27. Es de especial relevancia reseñar que la EMT es potencialmente reversible y, por tanto, las células mesenquimales podrían pasar de nuevo a células epiteliales MET (mesenchymal epitelial transition). Tanto la EMT como la MET son de gran interés y estudio en oncología, y más recientemente en otras disciplinas al ser una vía fundamental en el desarrollo de la fibrosis progresiva que lleva a un órgano al fallo funcional26,27. Numerosos estudios han demostrado cómo la familia de mi-ARN-200 tiene un papel central en la regulación de la EMT. Un estado de hipermetilación de las islas CpG cercanas al mi-ARN-200 favorecería la transición EMT, como se ha demostrado en la transformación en células mesenquimales. Además, la hipermetilación de estas islas se vería favorecida por modificaciones permisivas a nivel de histonas27,28. Por tanto, un tratamiento basado en la restitución de estas alteraciones podría reconstituir la expresión normal de los genes anormalmente expresados y devolver a la célula su fenotipo epitelial inicial. De hecho, en oncología el empleo de inhibidores de la ADN metiltransferasa e inhibidores de la acetilasa de histonas ha conseguido resultados muy esperanzadores en neoplasias de origen hematológico27.
Una característica distintiva de la fibrosis es que la cicatrización activa no cesa una vez que la lesión inicial ha sido contenida convirtiéndose en un proceso continuo. Los fibroblastos continuarían activos perpetuando el aumento de matriz y fibrosis progresiva. Así, fibroblastos aislados de pacientes con fibrosis renal permanecen activos, incluso cuando se cultivan in vitro29.
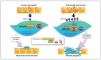
Se ha demostrado cómo esta actividad mantenida del fibroblasto es causada en parte por una hipermetilación del gen RASAL1, que codifica un inhibidor de la oncoproteína RAS, provocando una permanente activación fibroblástica, que estaría mediada por la expresión de este último29. Esta metilación estaría promovida por el TGF-β1 y mediada por la DNMT1. Estos cambios epigenéticos son estables y heredados a través de las sucesivas divisiones celulares. Por tanto, una exposición puntual del fibroblasto al TGF-β1 causaría una supresión puntual, reversible y sin metilación de RASAL1, mientras que una exposición más prolongada al TGF-β1 provocaría una metilación del RASAL1 induciendo una supresión transcripcional irreversible. La primera situación sería la observada en procesos fisiológicos de reparación, mientras que la última situación sería la observada en los procesos de fibrogénesis29 (fig. 3).

Figura 3. Esquema de la reparación del daño tisular y cambios epigenéticos. Cuando la lesión es de corta duración se secretan modestas cantidades de TGF-β1, que inducen la proliferación de fibroblastos y la secreción de matriz extracelular. En ausencia de TGF-β1, los fibroblastos regresan rápidamente o entran en apoptosis. En casos de lesión permanente o repetida, la producción de TGF-β1 es mucho mayor y mantenida provocando la metilación del ADN a nivel del promotor RASAL1, que conduce a una activación del sistema renina-angiotensina (SRA) de forma persistente, induciendo la proliferación y activación fibroblástica mantenida. Estas modificaciones son estables y se pueden heredar a través de múltiples divisiones celulares. La hidralacina, al favorecer la expresión de enzimas TET3, facilitaría la hidroximetilación de los residuos metilados de la citosina, promoviendo la transcripción de RASAL1 e inhibiendo la fibrogénesis. SRA: regulador muy importante que controla diferentes fenómenos, entre ellos la proliferación, la diferenciación, la adhesión y la migración celular. RASAL1: RAS protein activator like 1, actúa como supresor del SRA. TET3: enzima de la familia ten-eleven-translocation, que permite la demetilación del ADN.
La BMP7 (bone morphogenic protein) es una conocida proteína antifibrótica inhibidora del TGF-β1 y se ha comprobado su eficacia en modelos de fibrosis de ERC30. Estudios recientes muestran cómo la acción de la BMP7 estaría mediada por la desmetilación del RASAL1 a través de la hidroximetilación mediada por TET3. Es más, en situaciones de disminución de expresión de TET3, la BMP7 no desarrollaría su acción antifibrótica. Este hallazgo es trascendente, ya que en varias nefropatías se ha demostrado la disminución de TET3 (ND, nefropatía IgA, nefroesclerois hipertensiva o nefropatía lúpica)17.
La constatación de la importancia de las respuestas epigenéticas en el desarrollo de la fibrosis de la ERC abre nuevas posibilidades terapéuticas. Por un lado, la valoración en sangre periférica de fragmentos metilados CpG de los promotores de RASAL1 podría ser un biomarcador excelente del grado de fibrosis renal. Por otro, dado que la alteración en la metilación observada en el RASAL1 puede ser reversible, el uso de fármacos “epigenéticos” con capacidad de desmetilación podría ser un abordaje novedoso y eficaz. En la actualidad existen ya fármacos con capacidad de desmetilación y que se han utilizado en oncología, como el análogo nucleósido 5-azacitidina o la decitabina que inhiben la progresión de la fibrosis en varios órganos, incluidos riñón y corazón. Su elevada toxicidad hace improbable su utilidad en la enfermedad renal. Muy recientemente se ha descrito cómo la hidralacina induciendo la vía TET3 provocaría una desmetilación del ADN, que es similar a la capacidad de desmetilación de la 5-azacitidina y sin sus efectos genotóxicos. Además, su acción antifibrótica óptima se lograría a dosis bajas (25-50 mg/día), dosis inferiores a las utilizadas como antihipertensivo. Esta y otras terapias “epigenéticas” combinadas podrían mejorar el pronóstico de las enfermedades renales31,32 (fig. 3).
Epigenética y nefropatía diabética
En la ND, todos los tipos celulares del riñón se ven afectados, incluyendo podocitos, células mesangiales y endoteliales, el epitelio tubular, los fibroblastos intersticiales y el endotelio vascular. Valores elevados de glucosa inducen la expresión de factores de crecimiento como el TGF-β, la angiotensina II, los AGE (advanced-glycation end-products) y las citocinas inflamatorias, promoviendo la inflamación, fibrosis e hipertrofia, que contribuyen a la progresión de la ND. Estos factores, mediante la unión a sus receptores celulares, activarán factores de transcripción como Smads y NF-κB que promoverán la expresión de genes inflamatorios y profibróticos (revisado en referencia 33). Estos mecanismos alterarán el perfil epigenético en sus diferentes vías, como la metilación del ADN, la modificación de las histonas y la de la estructura de la cromatina, que a su vez aumentará y mantendrá la expresión de los genes inflamatorios y profibróticos. Además, recientemente los mi-ARN han demostrado tener un papel primordial en la regulación de los diferentes genes implicados en la ND34,35 (fig. 4).
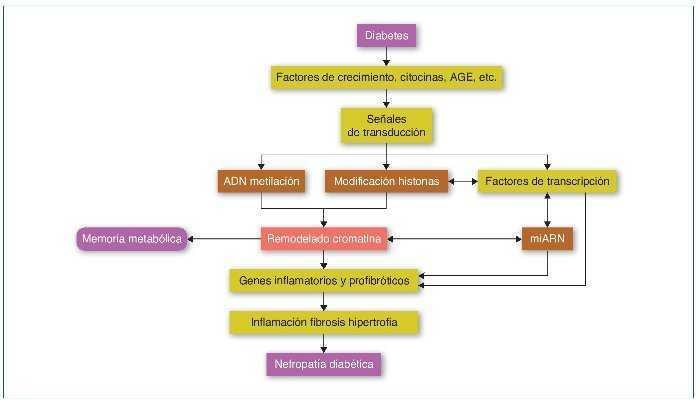
Figura 4. Modificaciones epigenéticas en la diabetes y su contribución en el desarrollo de la memoria metabólica y de la nefropatía diabética. AGE: advanced-glycation end-products. Modificada de referencia 35.
La progresión a largo plazo de complicaciones diabéticas como la ND, a pesar de un buen control glucémico, podría deberse a una memoria de la exposición inicial de las células diana a altos valores de glucosa, lo que conduciría a la persistencia de sus efectos deletéreos mucho después de la normalización de la glucosa; este fenómeno es conocido como memoria metabólica. Se piensa que mecanismos epigenéticos podrían estar implicados en este fenómeno36. Un estudio realizado en sangre periférica de pacientes pertenecientes a la cohorte DCCT/EDIC, que comparaba pacientes con progresión de retinopatía y nefropatía frente a grupo control sin progresión, mostró, en monocitos, un significativo aumento de la acetilación de la histona H3K9Ac de los genes relacionados con la inflamación que, además, se asoció significativamente con la media de HbA1c (hemoglobina glucosilada) a lo largo del seguimiento. Estos resultados, que sugieren una asociación entre HbA1c y la modificación de histonas, explicarían, al menos en parte y por mecanismos epigenéticos, la memoria metabólica36,37. Recientemente, estudios in vitro realizados en cultivos de células mesangiales han demostrado cómo la glucosa es un importante inductor de modificaciones de histonas que, al contribuir a la expresión de genes proinflamatorios, podría predisponer a la ND38.
Diferentes patrones de metilación de ADN en genes proinflamatorios se han puesto de manifiesto en pacientes con ND comparándolos con controles, tanto a nivel de sangre periférica como en saliva. Estos diferentes patrones de metilación de ADN, en especial en regiones potenciadoras de genes relacionados con la fibrosis, también se han demostrado estudiando células tubulares obtenidas por microdisección en pacientes con ERC (con y sin ND), y comparándolas con controles35.
Numerosos miARN se han asociado con el desarrollo de la ND, incluidos los de la familia miARN-200 y su papel regulador en el proceso de la EMT, ya comentado28,35,39. Los miARN estarían implicados tanto en promover como en atenuar la progresión de la ND. Recientemente se ha descrito en un modelo experimental de ND, como los valores de miARN-93 (disminuidos en la ND) provocarían cambios en la estructura de la cromatina. La diana del miARN-93 sería una cinasa Msk2 que induciría una fosforilización en la serina de la histona H3 (H3S10), que modificando la forma del nucleosoma favorecería la transcripción de numerosos genes relacionados con la ND. Valores altos de miARN-93 suprimirían la translación de la MsK2 mientras que una disminución en su expresión aumentaría los valores de Msk2. Esta observación es relevante, ya que la manipulación de un único miARN y sus acciones sobre la estructura del nucleosoma permitiría actuar sobre una amplia expresión de genes implicados en la ND. Por otra parte, el uso de inhibidores de la cinasa MsK2 podría tener claras implicaciones terapéuticas aún por explorar40.
EPIGENÉTICA Y PROTEINURIA
En los procesos que cursan con proteinuria, un hallazgo habitual es la presencia de marcadas alteraciones del podocito que se caracterizan por el borrado/fusión de los procesos podocitarios. Estas alteraciones estructurales podocitarias se han puesto en relación con una expresión disminuida del gen de la nefrina41.
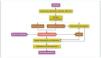
Recientemente se ha descrito cómo el factor de transcripción Kruppel-like KLF4, uno de los factores de transcripción con capacidad para reprogramar las células somáticas en células pluripotenciales, se expresa en la célula podocitaria. En condiciones normales permite un estado de hipometilación del promotor del gen de la nefrina. En su estado hipometilado, el promotor de la nefrina es activo y la nefrina se expresa con normalidad42. En la nefropatía con proteinuria, la expresión de KLF4 está disminuida, provocando una metilación del promotor de la nefrina, disminuyendo su expresión facilitando los cambios podocitarios y la proteinuria. La angiotensina II es uno de los mediadores responsables de esta menor expresión de KLF4. Se ha demostrado cómo el tratamiento mediante bloqueadores del receptor de la angiotensina es capaz de restaurar la expresión de KLF4 disminuyendo la metilación del promotor de la nefrina, aumentando su expresión y provocando un descenso en la proteinuria43,44 (fig. 5).
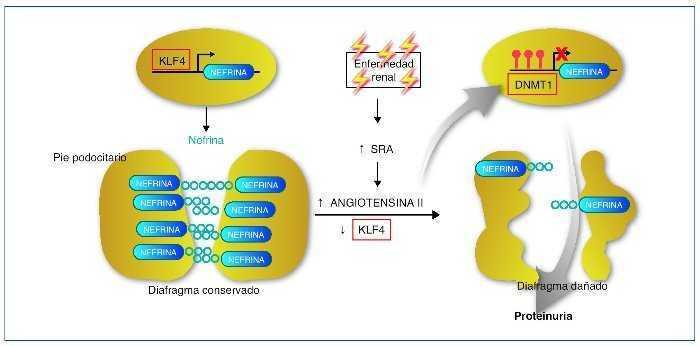
Figura 5. Mecanismo epigenético de la generación de proteinuria. En condiciones normales, el factor Kruppel-like (KLF4) se une al promotor del gen de la nefrina e impide la acción de la ADN metiltransferasa (DNMT1). Esto produce hipometilación de las CpG del promotor, lo que permite una normal expresión de la nefrina. La nefrina mantiene una estructura podocitaria adecuada y un diagrama podocitario eficiente, que no permitirá el paso de proteínas a la orina. En casos de enfermedad renal, un aumento de actividad del sistema renina-angiotensina (SRA) conducirá a un aumento de la formación de angiotensina II, que provocará una regulación negativa del KLF4. Esto permitirá la metilación de las CpG del promotor de la nefrina por medio de la DNMT1. La hipermetilación del promotor de la nefrina disminuirá su transcripción evitando su expresión. La expresión más baja de la nefrina contribuirá a una alteración podocitaria y de la hendidura podocitaria que provocará la proteinuria. Modificada de referencia 44.
En resumen, estos interesantes estudios demuestran un mecanismo de control epigenético en la diferenciación podocitaria y en el desarrollo de la proteinuria. Además, la activación del sistema renina-angiotensina (SRA) (vía común en diferentes tipos de nefropatías) afecta a esta diferenciación y provoca proteinuria a través de un “reseteo” epigenético. Estos resultados demuestran un nuevo mecanismo de acción de los bloqueadores del SRA, a través de la modulación epigenética, y abren nuevas dianas terapéuticas como las relacionadas con la metilación del ADN o bien con la inducción de factores de transcripción como el KLF444.
CONCLUSIÓN
La epigenética es una disciplina dinámica, que impulsa nuevos avances tecnológicos, así como desafiando y revisando los paradigmas tradicionales de la biología. A través de la epigenética se están empezando a comprender los roles y la interacción del ADN, el ARN y las proteínas, las relaciones entre el medio ambiente y la herencia y la etiología de la enfermedad. Se prevé que el campo de la epigenética contribuirá a la comprensión de las complejidades de la regulación genética, la diferenciación celular, la embriología, el envejecimiento y la enfermedad, pero también permitirá explorar sistemáticamente nuevas vías terapéuticas que lleven a la medicina personalizada.
En la nefrología, la epigenética contribuirá, al menos por 2 vías, a la mejor comprensión de la enfermedad renal. En primer lugar, el conjunto de conocimiento generado y el que está por venir, dilucidarán los mecanismos de la enfermedad renal como nunca se había sospechado, no solo a nivel molecular de los genes implicados sino, lo que es más importante, cómo estos genes son regulados y su posibilidad de modificación, explicando la variación fenotípica y la plasticidad de la enfermedad. En segundo lugar, la biología química epigenética y el descubrimiento de nuevos fármacos moduladores de la epigenética, permitirán un nuevo abordaje de la enfermedad renal. Aunque hay mucho que aprender en términos de mecanismos, utilidad terapéutica, eficacia y seguridad de fármacos dirigidos a modificadores epigenéticos, estos nuevos enfoques son prometedores para el futuro desarrollo de nuevas opciones de tratamiento. Apenas hemos arañado la superficie de las respuestas epigenéticas, todavía hay mucho que no sabemos y queda mucho más para aprender, pero ya no hay marcha atrás.
Agradecimientos
Mi especial agradecimiento al Dr. Mario F. Fraga, por sus correcciones y aclaración de conceptos.
Conflictos de interés
El autor declara que no tiene conflictos de interés potenciales relacionados con los contenidos de este artículo.
Conceptos clave
1. La epigenética se define como el estudio de las modificaciones genéticas estables que resultan en cambios de la expresión y función génica, sin una alteración correspondiente en la secuencia de ADN.
2. Los mecanismos de estas modificaciones comprenden la metilación en el ADN, la modificación de las histonas y los mi-ARN.
3. Investigaciones en este campo han evidenciado una estrecha relación entre modificaciones epigenéticas y el desarrollo de enfermedades.
4. Aunque las modificaciones epigenéticas son estables, son también susceptibles de ser modificadas, lo que abre un amplio y novedoso horizonte de posibilidades terapéuticas.
5. En nefrología, si bien su estudio es muy reciente, numerosos cambios epigenéticos ya han sido demostrados en el desarrollo y progresión de la ERC, la ND y el mecanismo de desarrollo de la proteinuria.
Correspondencia:
Miguel De La Torre
Sección de Nefrología. Hospital Universitario de Cabueñes.
C/ Los Prados, 395.
33394 Gijón.
mdtorre@senefro.org


