



Neurofibromatosis type 1 (NF1) is an autosomal dominant disorder that affects multiple organ systems.1 It can be diagnosed if a patient presents with two or more of the following features: six or more café-au-lait macules of larger than 5mm in diameter before puberty or larger than 1.5mm in diameter after puberty, axillary or inguinal skinfold freckling, two or more dermal neurofibromas or one plexiform neurofibroma, two ore more iris hamartomas, an optic pathway glioma, a distinctive long bone dysplasia involving the sphenoid wing or thinning of the long bone cortex with or without pseudarthrosis, and a first-degree relative with NF1.2 Neurofibromas, one of the main clinical features, usually present as discrete nodules or pedunculated masses, but cannot be presented as vasculitic skin purpura.3 Here, we report a female patient with NF1 who demonstrated vasculitic skin rash.
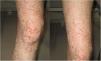
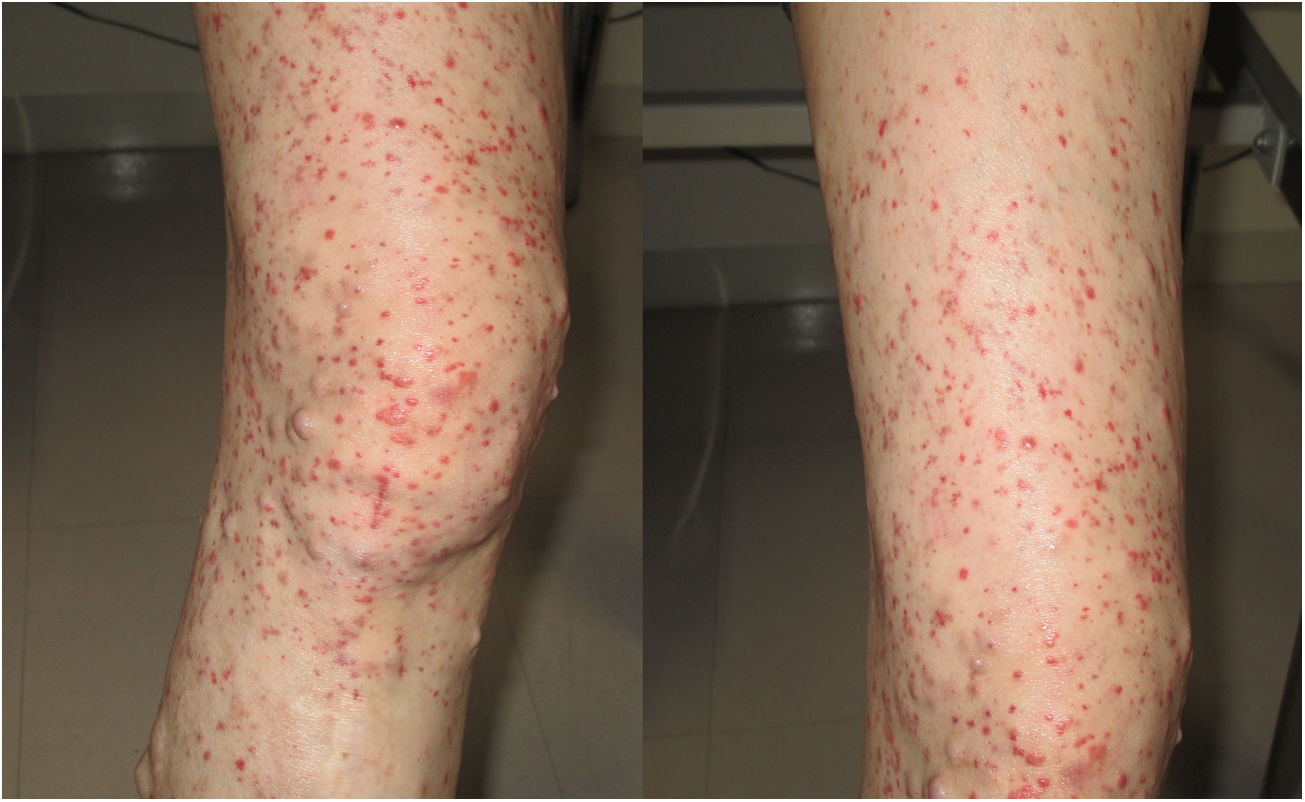
A 56-year-old Japanese women with neurofibromatosis type 1 presented with new onset of microscopic hematuria, abdominal pain, and arthralgia. Her past medical history included the left limb amputation due to a traffic accident and malignant peripheral nerve sheath tumours. She had no previous history of renal disease and denied any use of tobacco or alcohol. Upon physical examination, her respiratory rate was 14 breaths per minute, heart rate was 106 beats per minute, blood pressure was 137/85mmHg, and body temperature was 36.7°C. Notable examination findings included generalized neurofibromas, palpable purpura on the right leg (Fig. 1), and a slight tenderness at the epigastric area. The musculoskeletal examination showed mild pain in the bilateral elbow joints. Urinalysis showed >100 erythrocytes per high-power field and 0.54g/gCr of proteinuria. The kidney function was normal (blood urea nitrogen level, 15.6mg/dL; serum creatinine level, 0.44mg/dL). Renal biopsy revealed mild mesangial proliferation with granular Immunoglobulin A (IgA) deposition, and skin biopsy showed leukocytoclastic vasculitis with IgA deposition. The diagnosis of IgA vasculitis (IgAV) was made based on the European League Against Rheumatism, the Paediatric Rheumatology International Trials Organizations, and the Paediatric Rheumatology European Society (EULAR/PRNTO/PRES) classification criteria.4
IgAV is a small-vessel vasculitis, involving the skin, joints, and kidney. According to the EULAR/PRINTO/PRES classification criteria, the diagnosis of IgAV is confirmed by the presence of purpura and one of the following clinical manifestations: abdominal pain, arthralgia, renal insufficiency, and leukocytoclastic vasculitis with predominant IgA deposits.4 Proteinuria, hematuria, or renal insufficiency are present in 70–80% of adult patients with IgAV.5 There is no specific treatment for IgAV because it generally resolves spontaneously; however, renin–angiotensin system inhibitors and corticosteroid therapy are recommended to reduce proteinuria and maintain kidney function in moderate and severe cases.6
By contrast, neurofibromatosis type 1, an autosomal dominant disorder caused by germline mutations in the NF1 tumour suppressor gene, can manifest progressive multiple organ dysfunctions in the skin, bones, eyes, and neuropsychiatric system.1 Neurofibromas and café-au-lait macules are the main skin features, whereas, palpable purpura is not.3 Regular assessment of neurofibromas (owing to the increased risk of malignant peripheral nerve sheath tumours), vitamin D supplementation (owing to the increased risk of osteoporosis), blood pressure monitoring (owing to the increased risk of hypertension) and visual assessment (owing to the risk of optic pathway gliomas), are the mainstay of clinical management. Of note, patients with neurofibromatosis type 1 have the increased risk of developing malignancy such as brain tumours, adrenal cancer, and early-onset breast cancer.1
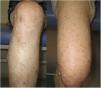
Although the association between IgAV and neurofibromatosis type 1 is no clear, this was the first reported case of IgAV in a patient with neurofibromatosis type 1. The esophagogastroduodenoscopy showed no evidence of gastrointestinal bleeding or gastrointestinal stromal tumours. After one month initiating enalapril, the patient's proteinuria and hematuria resolved, and the purpura subsided (Fig. 2). We recommended performing mammography for breast cancer screening because women with neurofibromatosis type 1 have the increased risk of breast cancer.1
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.



