









La glomerulopatía C3 es una entidad clinicopatológica definida por la presencia exclusiva o dominante de C3 en la inmunofluorescencia directa. Según el tipo y localización de los depósitos en la microscopia electrónica se distinguen dos subtipos: la glomerulonefritis C3 y la enfermedad por depósitos densos. La base fisiopatogénica de la enfermedad se encuentra en una alteración inmunológica o genética a nivel de las proteínas reguladoras del complemento, que conduce a una hiperactividad de la vía alternativa del complemento. Clínicamente se manifiestan como proteinuria, generalmente nefrótica, microhematuria y un grado variable de insuficiencia renal. El pronóstico renal es malo, alcanzando la insuficiencia renal crónica terminal hasta en un 60 % de los casos a los 10 años del diagnóstico. El tratamiento, aunque todavía no bien caracterizado, deberá ir dirigido a eliminar autoanticuerpos frente a las proteínas reguladoras del complemento, a restablecer dichas proteínas reguladoras deficientes o disfuncionantes o a eliminar proteínas mutantes o híbridas, así como los productos de degradación de C3b. Tras el trasplante, la recidiva de la enfermedad ocurre en el 65-70 % de los casos, con una pérdida de la función del injerto por este motivo en el 50 % después de 30-80 meses del trasplante.
DEFINICIÓN
La glomerulopatía C3 (GC3) es una enfermedad renal descrita de forma relativamente reciente y considerada como una patología primaria del sistema del complemento, en la que existe una desregulación de la vía alternativa que resulta en una hiperactivación de esta. Su definición viene dada por la presencia de un depósito mayoritario de C3 en ausencia de depósitos importantes de inmunoglobulinas en la inmunofluorescencia directa (IFD). En cuanto al patrón histológico en la microscopia óptica, inicialmente la glomerulonefritis membranoproliferativa (GNMP) fue el patrón histológico aceptado, lo que llevó incluso a una reclasificación de esta entidad. Pero la aparición de casos de glomerulonefritis necrotizantes, mesangiales o glomerulosclerosis segmentaria y focal con depósitos exclusivos de C3 en la inmunofluorescencia y una alteración en la vía alterna del complemento como base fisiopatogénica, hicieron que se modificara también esta parte de la definición.
Actualmente se reconocen dos formas de expresión en la GC3, la enfermedad por depósitos densos (EDD) y las glomerulonefritis C3 (GNC3), cuya definición va a ser determinada por los depósitos en la microscopia electrónica.
FISIOPATOGENIA
La patogenia de la GC3 se debe a una activación anormal en la vía alternativa del complemento, fundamentalmente a nivel sérico (fase fluida), que conducirá a una producción excesiva de C3 activa (C3b) y sus productos de degradación (iC3b y C3dg), que se depositarán en el glomérulo.
Esta activación excesiva de la vía alternativa del complemento puede ser debida a dos causas fundamentalmente: a) mutaciones en las proteínas reguladoras de la vía alternativa del complemento o, más frecuentemente, b) presencia de autoanticuerpos frente a estas.
Entre los autoanticuerpos, el más frecuente y más ampliamente descrito en la literatura es el factor nefrítico (C3NeF). C3NeF es un anticuerpo que se une a la C3 convertasa de la vía alternativa (C3bBb) e impide su disociación espontánea, estabilizando su función. Esto es, C3NeF mantiene activa a la C3 convertasa de la vía alternativa, de manera que no deja de escindir C3 en C3a y C3b y, como consecuencia de ello, conduce a un consumo masivo de C3 (C3 sérico estará disminuido), imposibilitando la acción de las proteínas reguladoras sobre dicho complejo enzimático. C3NeF está presente hasta en un 80 % de los casos de EDD y hasta en un 46 % de las GNC31-3. Otros anticuerpos cuya forma de actuación es similar a C3NeF, dado que impiden la disociación de la C3 convertasa, son los anticuerpos anti-FB y anti-C3b, que reconocerán de forma aislada a estos componentes de la convertasa, Bb o FB y C3b, respectivamente. Son extremadamente raros, descritos únicamente en casos aislados4,5 (figura 1). El último de los autoanticuerpos descritos en la GC3, mucho más frecuente en casos de síndrome hemolítico urémico (SHU), es el anticuerpo anti-FH. El factor H (CFH) es la principal proteína reguladora de la vía alternativa del complemento. Su actividad reguladora es ejercida tanto en la fase fluida (actúa como cofactor para inactivar C3b, así como en la disociación de la C3 convertasa) como en la superficie celular, previniendo la activación de la vía y, por tanto, la formación del complejo de ataque de membrana (membrane atack complex: MAC, C5b-9). CFH es una proteína soluble formada por 20 dominios (figura 2). En el extremo N-terminal (short consensus repeat, SCR 1-4) se encuentra la región que actúa como cofactor para la inactivación de C3b, así como la que promueve la disociación de la C3 convertasa. Sin embargo, el extremo C-terminal (SCR 19-20) es el responsable del reconocimiento de las superficies celulares. Por tanto, no es difícil inferir de esto que los anticuerpos anti-FH que vayan dirigidos frente a la región SCR 19-20 (C-terminal) favorecerán el desarrollo de SHU, mientras que los que se dirijan frente a los dominios SCR 1-4, darán lugar a un defecto en la regulación de C3 en fase fluida y, por tanto, originarán una GC36-10. Esto mismo ocurrirá en caso de tratarse de una mutación en el CFH.
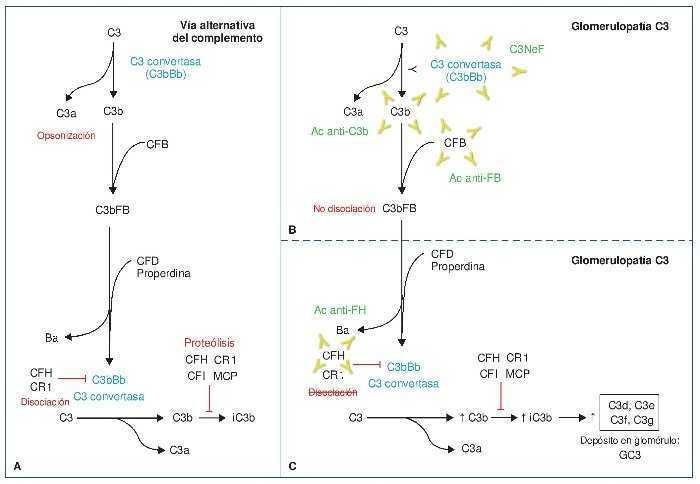
Figura 1. Vía alternativa del complemento.
A) Vía alternativa del complemento. Escisión de C3 en C3a (anafilatoxina) y C3b. C3b se une a la fracción b del factor B (Bb) para dar lugar a la C3 convertasa (amplificación de la vía), que a su vez generará más C3b. La escisión del factor B (FB) en su fracción a (Ba) y su fracción b (Bb) está regulada por el factor D y la properdina. C3b, además de lo descrito, será inactivado por FH y CR1 (iC3b) y degradado a C3c y C3dg. B) Glomerulopatía C3 mediada por anticuerpos (Ac) estabilizadores. Por un lado, C3NeF (factor nefrítico), el más frecuente, es un anticuerpo que estabiliza a la C3 convertasa, de manera que no puede ser inactivada por FH. Dará lugar a una producción descontrolada de C3b. Por otro lado, Ac frente a Bb y C3b, que también estabilizarán las dos moléculas, impidiendo su disociación. De esta manera, la C3 convertasa (C3bBb) no dejará de funcionar, dando de nuevo lugar a una producción descontrolada de C3b. C) Glomerulopatía C3 mediada por Ac anti-FH. Los AC anti-FH inhiben la acción reguladora que FH ejerce inactivando la C3 convertasa, así como su acción de inactivación de C3b, por lo que nuevamente daría lugar a una producción descontrolada de C3b que, además, no es inactivado.
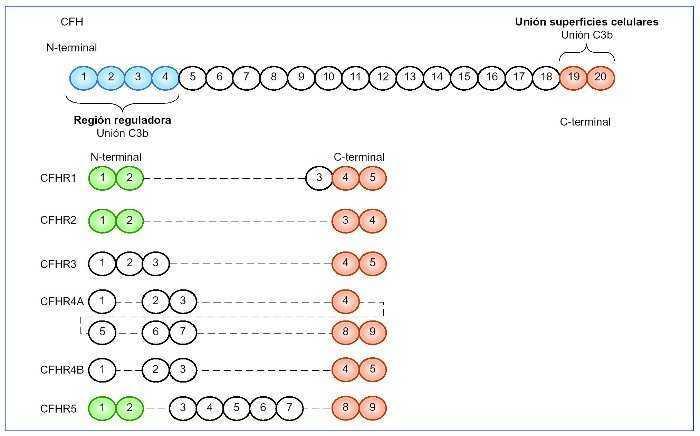
Figura 2. FH y factores relacionados.
El FH está compuesto por 20 dominios (SCR), algunos de los cuales son muy importantes. En el N-terminal se encuentran los dominios 1-4 encargados de la función reguladora de la proteína. Reconocen a C3b a nivel sérico (fase fluida de la vía alternativa del complemento). En el C-terminal se encuentran los dominios 19 y 20, cuya función principal es el reconocimiento de C3b unido en las superficies celulares, de manera que reconocerá si este se encuentra sobre la superficie de una célula huésped o patógena. Los dominios del extremo C-terminal del CFH (en naranja) están muy conservados en todos los CFHR, de manera que estas proteínas relacionadas con CFH mantienen la capacidad de reconocimiento de C3b unido a la superficie celular. Sin embargo, los dominios del extremo N-terminal (en azul) no se encuentran en los CFHR, por lo que no poseen la actividad reguladora de CFH. Los SCR 1-2 (en verde) están muy conservados en CFHR1, CFHR2 y CFHR5, muy importantes en la dimerización.
Por otro lado, mutaciones en los genes que codifican las proteínas reguladoras del complemento también podrán conducir a una activación excesiva de la vía alternativa del complemento en fase fluida, lo que conducirá al desarrollo de una GC3. Como hemos visto anteriormente, mutaciones en la región N-terminal del CFH favorecerán el desarrollo de una GC33,11-13, aunque también si se localizan fuera de esta región y dan lugar a una deficiencia parcial o completa de la proteína3,12,14-18. Además se han descrito cinco proteínas que muestran una secuencia y estructura homóloga a CFH, llamadas proteínas relacionadas con CFH 1-5 (CFHR 1-5) (figura 2). Su función no es del todo conocida, aunque parece que CFHR1, CFHR2 y CFHR3 participarían inhibiendo la actividad del complemento a nivel de la C3 convertasa y de la C5 convertasa19-21, y CFHR5 únicamente bloquearía la actividad de la C3 convertasa22. Los dominios SCR 1-2 están muy conservados en CFHR1, CFHR2 y CFHR5 y son importantes para formar homo o heterodímeros23. Estos dímeros tendrían una mayor afinidad por C3b, de manera que competirían con CFH en esta unión, e impedirían la acción reguladora de este desencadenando un aumento no controlado de C3b y sus productos de degradación, que se depositarían en la membrana basal glomerular (MBG), dando lugar a una GC323. Este mecanismo es el que se ha visto asociado a las mutaciones, duplicaciones internas o formación de genes híbridos descritas en los CFHR. Gale et al24,25 describieron en 2010 la “nefropatía chipriota”, en la que existe una duplicación interna de los dominios SCR 1-2 del CFHR5. Más tarde, en 2014, Medjeral- Thomas et al26 demostraron que otra mutación en heterocigosis en CFHR5 daba lugar a la misma duplicación interna de los dominios SCR 1-2 de dicha proteína, que ya se había observado en la nefropatía chipriota, pero en esta ocasión en una familia sin antecesores chipriotas, de manera que pasó a llamarse la “nefropatía CFHR5”. En 2012, Malik et al27 describen un gen híbrido, CFHR3-CFHR1, como consecuencia de una deleción del genoma que compromete a los exones 4-6 de CFHR3 y al exón 1 de CFHR1, de manera que se produce una proteína híbrida en la que los dominios SCR 1 y 2 de CFHR3 se unen a la proteína CFHR1 completa. En 2013, Tortajada et al28 descubren una mutación que provoca la duplicación de los SCR 1-4 de CFHR1, dando lugar a una proteína mutante capaz de formar homo y heterooligómeros con CFHR1, CFHR2 y CFRH5. Otra proteína híbrida descrita en 2014 por Chen et al29 es la proteína CFHR2-CFHR5, generada como consecuencia de una deleción en los exones 4-5 de CFHR2, de modo que se unirían los dominios SCR 1 y 2 de CFHR2 a la proteína CFHR5 completa (figura 3).

Figura 3. Alteraciones descritas en los factores relacionados con el factor H (CFHR) causantes de glomerulopatía C3.
A) Gen mutante CFHR5 con duplicación de SCR1 y 2, por duplicaciones en los exones 2 y 3 de CFHR524,26. B) Gen híbrido resultado de la unión de SCR1 y 2 de CFHR3 a CFHR1 completa, como resultado de las deleciones en los exones 4-6 de CFHR3 y el exón 1 de CFHR127. C) Gen mutante CFHR1 con duplicación de SCR1-4, por mutación en los exones 2-5 de CFHR128. D) Gen híbrido resultado de la unión de SCR1 y 2 de CFHR2 a CFHR5 completa debido a deleciones en los exones 4 y 5 de CFHR229.
Además de las anormalidades genéticas en los genes CFH y CFHR, también se han descritos mutaciones en otros genes del complemento que conducirán al desarrollo de una GC3, aunque con una frecuencia muy baja (el 17 y el 20 % en GC3 y EDD, respectivamente)3. Por un lado pueden ser mutaciones que conlleven una hiperactivación de la vía alternativa del complemento como consecuencia de la ganancia de función de CFB y C330-32 o, por el contrario, por pérdida de función de las proteínas reguladoras CFI y MCP3 (como ocurre con las mutaciones de CFH). Por tanto, tras el diagnóstico de una GC3 sería recomendable realizar un estudio completo del complemento, para conocer exactamente la causa fisiopatogénica de la enfermedad (tabla 1).

PRESENTACIÓN CLÍNICA
El órgano principalmente afectado en esta entidad, como su propio nombre indica, es el riñón. La forma de presentación es muy variable y puede manifestarse como síndrome nefrótico hasta en el 30-50 % de los casos3,33,34, síndrome nefrítico e incluso anormalidades urinarias aisladas en forma de proteinuria no nefrótica o microhematuria. Esta alteración en el sedimento es prácticamente universal en esta entidad. La función renal en el momento del debut tampoco es constante, puesto que pueden presentar desde una función renal normal hasta un fracaso renal agudo grave en el contexto de un síndrome nefrítico. Sin embargo, lo habitual es un deterioro de la función renal leve, con un filtrado glomerular en torno a 65-70 ml/min/1,73 m2 3,33. La hipocomplementemia C3 es frecuente, pues está presente en el 45-60 %3,33, pero no es imprescindible para su diagnóstico, por lo que la ausencia de esta no debe descartar la enfermedad. Por el contrario, la presencia de C3 sérico bajo de forma aislada no sucede únicamente en esta patología glomerular, pues también puede estar presente en las GNMP por depósito de inmunocomplejos3, entre otras.
Las manifestaciones extrarrenales de las GC3, aunque poco frecuentes, deben ser conocidas. Todas ellas comparten con la enfermedad renal la base fisiopatogénica, es decir, están estrechamente asociadas a la regulación de las proteínas de la vía alternativa del complemento. La que probablemente es más conocida es el SHU. Esta microangiopatía trombótica puede aparecer antes35 o después3,36-40 del debut de la enfermedad glomerular, o incluso hacer su debut después de que el paciente es trasplantado tras alcanzar una insuficiencia renal crónica terminal por la GC338. Otra afectación extrarrenal de la enfermedad es la degeneración macular asociada a la edad. En esta enfermedad se han descrito factores genéticos relacionados con mutaciones en CFH41-43, aunque la asociación de las dos entidades es infrecuente. Lo que presentan estos pacientes es una pérdida progresiva de visión nocturna, objetivándose una atrofia retiniana con drusas y depósitos con o sin hemorragias43. Y la última de las afectaciones asociadas a las GC3 típicamente, aunque como las anteriores no es frecuente, es la lipodistrofia. La lipodistrofia se caracteriza por la pérdida progresiva y simétrica de grasa subcutánea a nivel facial, cuello, extremidades superiores, tórax y abdomen, en una secuencia cefalocaudal que respeta las extremidades inferiores. Aproximadamente, un 83 % de los pacientes con lipodistrofia tiene hipocomplementemia y positividad de C3NeF, y hasta el 22 % de los casos desarrolla una GNMP tras 8 años de seguimiento44.
HISTOLOGÍA
Como ya se ha comentado anteriormente, la histología va a ser crucial para el diagnóstico de la entidad, fundamentalmente la técnica de inmunofluorescencia. El patrón de microscopia óptica más habitual es el de una GNMP. De hecho, la definición de las GC3 condujo a la reclasificación de esta entidad histopatológica, de manera que dejaron de definirse por la distribución de los depósitos en la microscopia electrónica (antiguas GNMP tipo1, tipo 2 y tipo 3) y pasaron a definirse en función de la inmunofluorescencia (GNMP mediadas por complemento o GC3, y GNMP mediadas por inmunocomplejos)45. Sin embargo, tras la aparición de varios casos de patrones histológicos diferentes, como glomerulonefritis necrotizante con semilunas46-49, glomerulosclerosis segmentaria y focal50 y glomerulonefritis mesangiales49, con un depósito exclusivo de C3 en la IFD y una hiperactividad de la vía alternativa del complemento, la microscopia óptica perdió valor en el diagnóstico.
En cuanto a los depósitos en la IFD, inicialmente solo se incluían dentro de la entidad las glomerulopatías que tuvieran un depósito exclusivo de C3. No obstante, tras ser realizada una revisión de las biopsias de los pacientes con EDD, enfermedad gold standard dentro de las GC3, en la Columbia University, se observó que la mitad de los casos quedaba fuera de la definición. Sin embargo, con la definición de un depósito de C3 dominante, de manera que este fuera al menos dos magnitudes de intensidad superior al depósito de las inmunoglobulinas, se incluían casi el 90 % de los casos de EDD51. Es por este motivo por el que actualmente se acepta el depósito predominante de C3 en presencia de inmunoglobulinas, si el primero es al menos dos magnitudes de intensidad superior a las últimas en la IFD, como definición de la enfermedad. Los depósitos se localizan en el mesangio y/o a lo largo de las paredes capilares glomerulares.
La microscopia electrónica nos va a permitir distinguir entre los dos tipos o clases de GC3: a) por un lado estaría la EDD, en la que los depósitos ocupan completamente la lámina densa de la MBG y son de material muy denso, generalmente lineales y extensos, muy característicos en su visualización ultraestructural, y b) por otro lado se encontraría la GNC3, en la que los depósitos son más pequeños y menos electrón-densos que en la EDD, y pueden localizarse tanto subendoteliales como mesangiales, y menos frecuentemente subepiteliales (figura 4).
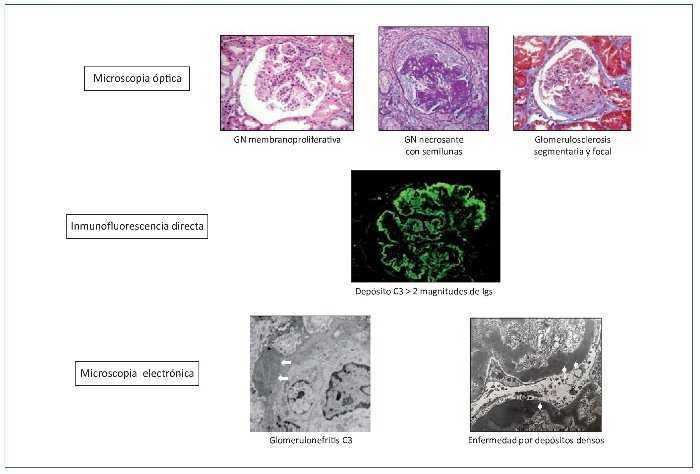
Figura 4. Definición de glomerulopatía C3.
Microscopia óptica: el patrón en la microscopia óptica no tiene valor para el diagnóstico de la enfermedad. No obstante, la glomerulonefritis (GN) membranoproliferativa es el patrón histológico más frecuente. Inmunofluorescencia directa: el diagnóstico viene dado por el depósito granular membranoso y/o mesangial aislado o predominante de C3 en la inmunofluorescencia directa. Microscopia electrónica: la distinción entre enfermedad por depósitos densos y glomerulonefritis C3 depende de la clase de depósitos en la microscopia electrónica. Depósitos subendoteliales y/o subepiteliales (flechas blancas) y mesangiales en la glomerulonefritis C3. Depósitos electrón-densos intramembranosos, lineales y extensos (puntas de flecha blancas) en la enfermedad por depósitos densos.
PRONÓSTICO
La supervivencia renal en esta enfermedad no es muy esperanzadora. Según las series3,52, entre el 40-60 % de los pacientes se encontraban en terapia renal sustitutiva tras 10 años de seguimiento. Sin embargo, en ninguna de estas series se ha podido demostrar que la GNC3 tuviera una supervivencia diferente de la EDD. Únicamente, en la serie de Servais et al3 se pudo demostrar una peor supervivencia renal a 10 años de la EDD, cuando la enfermedad debutaba a edad adulta. No obstante, en ninguna de estas dos series se diferencia la supervivencia entre los pacientes que recibieron tratamiento inmunosupresor específico y los que no lo recibieron. Este aspecto, que sí está evaluado en la serie de Rabasco et al33, es muy interesante. En la serie española se demostró que la supervivencia renal era mejor en los pacientes tratados con inmunosupresión, especialmente si estaba basada en micofenolato y esteroides, que en los que únicamente recibieron tratamiento antiproteinúrico para la enfermedad (supervivencia a 10 años del 100 frente al 70 %; p = 0,043). Incluso, a pesar de que los pacientes que recibieron inmunosupresión fueron los que debutaron con una mayor gravedad (síndrome nefrótico del 20 y el 67 % en pacientes no tratados frente a pacientes tratados, respectivamente).
Las cohortes de pacientes publicadas en esta entidad no son grandes debido a que se trata de una enfermedad rara y recientemente definida. Sin embargo se han evaluado algunos factores de mal pronóstico de progresión a insuficiencia renal3,52. Parece que el único factor hallado de forma común en el análisis multivariante en ambos trabajos es la edad de presentación. Cuanto mayor es el paciente en el momento del debut, peor es el pronóstico. Otros factores encontrados, aunque no confirmados por otros autores, son el diagnóstico de EDD frente a GNC3, la presencia de semilunas y un filtrado glomerular < 60 ml/min/1,73 m2 en el momento del diagnóstico. A pesar de que el grupo de Servais pudo demostrar en el análisis univariante que el uso de inhibidores de la enzima convertidora de angiotensina (IECA) se asociaba con mejor pronóstico renal, no fue capaz de demostrar que el tratamiento inmunosupresor también lo era, como parece concluirse en el trabajo de Rabasco et al.
TRATAMIENTO
El uso de IECA o antagonistas de los receptores de la angiotensina II en esta enfermedad se ha extrapolado dada su eficacia en otras enfermedades renales proteinúricas. Sin embargo, no hay una evidencia específica que apoye su uso en esta enfermedad, a excepción de los datos mostrados por la cohorte francesa3, en cuyo análisis univariante el uso de estos agentes estaría asociado a mejor pronóstico renal.
El objetivo de las terapias dirigidas al tratamiento de las GC3 debería ser: a) eliminar autoanticuerpos frente a las proteínas reguladoras del complemento, como C3NeF o anti-FH; b) restablecer proteínas reguladoras deficientes o disfuncionantes53,54, o c) eliminar proteínas mutantes o híbridas, así como los productos de degradación de C3b (iC3b, C3c y C3dg).
El tratamiento inmunosupresor basado en esteroides, micofenolato, ciclofosfamida, ciclosporina u otros antilinfocíticos, así como la plasmaféresis, estaría dirigido frente al primero de los objetivos enumerados, la eliminación de autoanticuerpos. Por tanto, los pacientes que tengan alguna anomalía genética en los genes que expresan las proteínas del complemento, probablemente no obtendrán ninguna respuesta a dicho tratamiento. Debido a esta heterogeneidad en la enfermedad, estos tratamientos se han considerado ineficaces3,52,56-61, aunque existe mucha controversia. Hay autores como D’Agati55 o el grupo español33 que sí defienden el uso de esteroides y/o micofenolato en las GC3. En la revisión del grupo de la Columbia University, los 18 pacientes que recibieron tratamiento inmunosupresor presentaban una albúmina sérica significativamente menor que los no tratados, así como un mayor porcentaje de semilunas en la biopsia renal, a pesar de lo cual aparentemente presentaron una mejor evolución, puesto que en el análisis univariante el tratamiento inmunosupresor tenía un efecto protector, aunque no alcanzó la significación estadística (p = 0,051), más eficaz si se asociaba a tratamiento antiproteinúrico. Asimismo, Rabasco et al33 encuentran una diferencia estadísticamente significativa con respecto a la llegada a insuficiencia renal crónica terminal, objetivo principal del estudio, entre los pacientes que reciben tratamiento inmunosupresor y los que no, a favor de los primeros (el 7 frente al 35 %; p = 0,012). Además, dentro del grupo de pacientes tratados con inmunosupresión, los que recibieron micofenolato y esteroides como base del tratamiento presentaron mayor remisión clínica y menor duplicación de la creatinina sérica que los tratados con otro tipo de inmunosupresión (remisión clínica del 86 frente al 50 %, p = 0,018; duplicación de creatinina de 0 frente al 37 %, p = 0,002). Sin embargo hay otros autores que rechazan su eficacia, tanto en la EDD58,59 como en la GNC33.
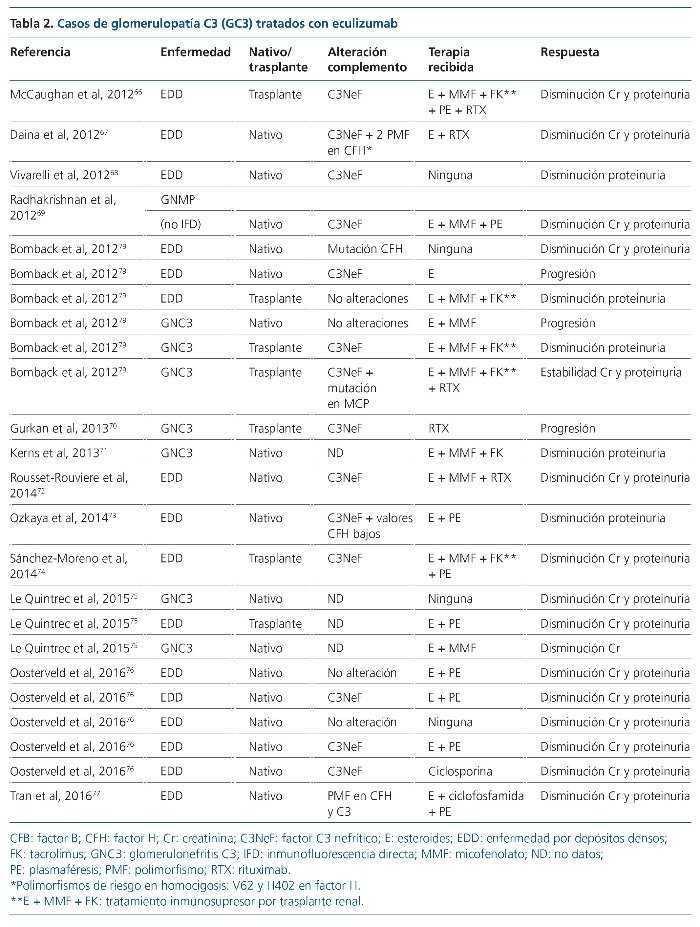
Las terapias que actúan directamente frente al complemento han comenzado a expandirse recientemente, tras el aumento del conocimiento de la base fisiopatogénica de la enfermedad. Actualmente están enfocadas fundamentalmente a la restauración de las proteínas reguladoras deficientes o disfuncionantes, así como dirigidas al bloqueo de la formación del complejo de ataque de membrana, cuya justificación y eficacia, como veremos más adelante, es difícil de explicar. El factor CR1 es una glucoproteína localizada en algunas superficies celulares (eritrocitos, monocitos, neutrófilos, linfocitos B, algunos linfocitos T, células dendríticas, podocitos)62-65 y modula la cascada del complemento a diferentes niveles (figura 5). Entre sus funciones se encuentra la de inhibir a la C3 convertasa, así como favorecer la inactivación de C3b a iC3b, y ser cofactor para la escisión de iC3b a C3c y C3dg. En un modelo animal con ratones knockout para el gen de CFH (Cfh–/–, deficiencia completa de esta proteína) que desarrollan una GC3, Zhang et al64 infunden por vía intravenosa o intraperitoneal CR1 soluble. Cuarenta y ocho horas tras la infusión repiten la biopsia renal, objetivando un aclaramiento casi completo de los depósitos de C3, así como un marcado aumento de C3 sérico. Después de la obtención de este esperanzador resultado se ha puesto en marcha un ensayo clínico fase I con esta molécula (sCR1, TP10), que ha finalizado, pendiente de resultados (NCT01791686, www.clinicaltrials. gov). Otra terapia, que todavía está en fase de experimentación, es el uso de una minimolécula de CFH. Nichols et al65 infunden por vía peritoneal una molécula recombinante de CFH, que únicamente contiene los dominios SCR esenciales de este (1-4 y 18-20), a unos ratones knock-out para CFH con GC3. Tras la infusión comprueban que los valores de C3 sérico aumentan y los depósitos de C3 en el glomérulo disminuyen. Estas dos terapias que actúan sobre la vía alternativa del complemento, lo hacen impidiendo o disminuyendo la formación de C3b y sus productos de degradación, moléculas que de no eliminarse irán a depositarse al glomérulo dando lugar a las GC3. Pero, ¿por qué el bloqueo de la formación del complejo de ataque de membrana también es eficaz en algunos casos de GC3? La experiencia de eculizumab en el tratamiento de GC3 se basa en la publicación de casos y series de casos66-77, así como en un ensayo clínico con seis pacientes78. De los 24 casos publicados (16 EDD y 8 GNC3), seis son niños y ocho son recidivas en el trasplante renal (tabla 2). Veinte (83 %) presentan respuesta al tratamiento con el anticuerpo monoclonal anti-C5, lo que evitó la diálisis crónica en tres de ellos. No obstante, en el único ensayo clínico que se ha realizado78, de los seis pacientes tratados, tres presentan una respuesta favorable, uno se mantiene estable y dos progresan hacia la insuficiencia renal crónica. Parece que todos los autores concluyen que, probablemente, la respuesta favorable a eculizumab tiene lugar en los pacientes en los que se demuestran valores séricos elevados del MAC (C5b-9) o cuando este está depositado en el glomérulo. De esta manera, el tratamiento con eculizumab bloquearía la formación del MAC y, por tanto, el daño que su depósito produce a nivel glomerular. Esto demuestra una vez más que la GC3 es una entidad heterogénea. Los casos en los que existe una elevación del MAC sérico y/o se encuentra depositado en el glomérulo podrían tener una mayor desregulación de la C5 convertasa que de la C3 convertasa, contrario a lo habitual en las GC380 y, consecuentemente, responderían favorablemente al tratamiento con eculizumab.

Figura 5. Terapias directas frente al complemento.
A) Factor soluble CR1, cuya eficacia en ratones ya fue probada por Zhang et al64. Actualmente ha finalizado un ensayo clínico fase I, pendiente de resultados, tanto en adultos como en niños con glomerulopatía C3 (www.clinicaltrials.gov). B) Miniproteína recombinante del factor H (mini-CFH) que contiene los dominios 1-4 y 18-20, ensayada con éxito en ratones por Nichols et al65. C) Eculizumab, anticuerpo monoclonal humanizado que se une con alta afinidad a C5 impidiendo la acción de la C5 convertasa sobre este y, por tanto, impidiendo la formación del complejo de ataque de membrana.
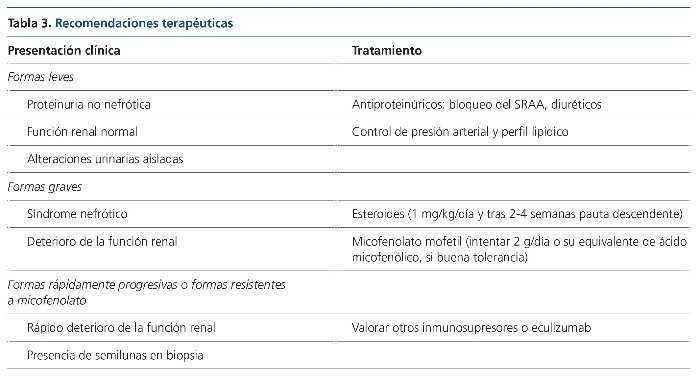
No obstante, la correlación fisiopatogenia-tratamiento no ha sido explorada como objetivo principal por ningún autor, si bien en la cohorte española ya se señala este hecho. En este trabajo se observa que el 80 % de los pacientes que presentaban C3NeF positivo y fueron tratados con tratamiento inmunosupresor presentaron remisión parcial o completa, mientras que solo tres de los ocho tratados con C3NeF negativo alcanzaron dicho objetivo, llegando a enfermedad renal crónica terminal un 20 frente a un 63 %, respectivamente. A la espera de estudios que exploren esta correlación, nuestro grupo emplea un protocolo de tratamiento según la tabla 3.
TRASPLANTE
El riesgo de recurrencia de la EDD está bien descrito en la bibliografía80-82, mientras que la evidencia en el caso de las GNC3 es mucho menor3,34,83. En la EDD existe un riesgo de recurrencia en torno al 70 %, y hasta un 50 % pierde el injerto por este motivo en los 30 meses siguientes al diagnóstico. Parece que el riesgo de recurrencia de la GNC3 es similar al de la EDD. Aproximadamente, dos tercios de los pacientes con GNC3 presentan recidiva de la enfermedad a los 28 meses del trasplante, con una pérdida del injerto por este motivo similar a la EDD (50 %), en una mediana de tiempo de 18 meses desde el diagnóstico de la recidiva83. Los factores de riesgo de recidiva no son bien conocidos, aunque algunos autores apuntan que la agresividad de la enfermedad en los riñones nativos podría ser determinante80. Otros factores como donantes de vivo frente a fallecido o el tipo de inmunosupresión en la inducción no tienen una clara asociación con el curso de la enfermedad postrasplante. Sin embargo, todos están de acuerdo en extremar la precaución si el donante de vivo está relacionado con el receptor, puesto que, extrapolando la experiencia con el SHU, la nefrectomía podría llevar al desarrollo de la enfermedad en el donante84. Otro grupo de mayor riesgo de recidiva, así como de menor tiempo hasta esta, serían los pacientes con una GNC3 en el contexto de una paraproteína monoclonal83.
La forma de presentación es similar a la presentada en los riñones nativos con proteinuria, generalmente nefrótica, y microhematuria con mayor o menor grado de deterioro de la función renal.
No hay ningún estudio que evalúe la asociación entre el tipo de alteración en la vía alternativa del complemento, adquirida o genética, y el riesgo de recidiva. Tampoco se ha estudiado si existe un mayor riesgo de recidiva en los pacientes que presentan positividad de autoanticuerpos frente a las proteínas de la vía alternativa del complemento (anticuerpos anti-FH y C3NeF) en el momento del trasplante, comparados con los que pudieran conseguir negativizarlos pretrasplante.
Lo que sí parece prudente, más aún si el donante va a ser un donante vivo emparentado, es realizar un estudio genético y funcional del complemento, para determinar la causa de la hiperactividad en la vía alternativa del complemento que lleva al desarrollo de la enfermedad (tabla 1). En el caso de la presencia de autoanticuerpos, la intensificación del tratamiento inmunosupresor basado en micofenolato33 u otro agente frente a linfocitos T y B, como rituximab84, podría retrasar o prevenir la recidiva. Sin embargo, en aquellos con alteraciones genéticas estas medidas probablemente sean ineficaces, pudiendo ensayarse otras terapias como la infusión de plasma fresco congelado (que reemplazaría el CFH deficiente)85 o el tratamiento con eculizumab u otros nuevos agentes reguladores como CR1 soluble, ya descrito previamente en esta revisión. Por supuesto, si la causa de la GC3 es la presencia de una paraproteína monoclonal, el tratamiento específico de esta podría ser beneficioso. De los pacientes con una GC3 y tratados con eculizumab detallados en la tabla 2, siete son trasplantados, cinco presentan buena respuesta, con disminución de proteinuria y creatinina, otro permanece estable sin progresión durante el año en el que está recibiendo eculizumab y otro progresa a insuficiencia renal crónica terminal. Como ocurre con la enfermedad en los riñones nativos, la respuesta a eculizumab es muy variable y, según los autores, probablemente relacionada con los valores séricos de C5b-9 o con su depósito en el glomérulo. El tratamiento posiblemente debería mantenerse durante un largo período, puesto que en el único ensayo clínico realizado79, tras la suspensión del fármaco después de 12 meses, dos de los cuatro pacientes que respondieron presentaron una nueva recidiva.
Conflictos de interés
M.P. declara haber recibido honorarios de Alexion Pharmaceuticals por dar charlas y participar como consejero.
T.C. declara que no tiene conflictos de interés potenciales relacionados con los contenidos de este artículo.
Conceptos clave
1. La GC3 se define por la inmunofluorescencia, independientemente del patrón histológico descrito en la microscopia óptica. La localización de los depósitos en la microscopia electrónica es importante para distinguir la GNC3 y la EDD.
2. Se manifiesta con microhematuria, distintos grados de insuficiencia renal y proteinuria que puede alcanzar el síndrome nefrótico. La hipocomplementemia C3 no es constante.
3. La base fisiopatogénica de la enfermedad es una hiperactividad de la vía alternativa del complemento, ya sea a nivel genético o a nivel inmunológico. Puesto que esto tiene implicaciones terapéuticas, es recomendable la realización de un estudio del complemento.
4. El tratamiento puede variar desde el uso de antiproteinúricos a tratamiento con inmunosupresores basados en micofenolato y esteroides. En las formas rápidamente progresivas o resistentes a inmunosupresión puede ensayarse el uso de eculizumab. Están en fase de estudio nuevos bloqueantes del complemento.
5. La recidiva en el trasplante renal es muy frecuente, con pérdida de la función del injerto hasta en un 50 % de los casos dentro de los 2-3 años posteriores al diagnóstico de la recidiva.
Correspondencia: Teresa Cavero Escribano
Servicio de Nefrología.
Hospital Universitario 12 de Octubre.
Avda. de Córdoba, s/n.
28041 Madrid.
tcaveroescribano@gmail.com


