


Fabry disease is a multisystem lysosomal storage disorder caused by mutations in the GLA gene that result in a deficient or absent activity of alpha-galactosidase A. There is a wide spectrum of GLA gene variants, some of which are described as non-pathogenic. The clinical importance of the D313Y variant is still under debate, although in recent years it has been considered as a variant of unknown significance or a benign variant. Despite this prevailing notion, there are multiple case reports of patients with D313Y variant that presented signs and symptoms consistent with FD without any other etiological explanation. In this article, we present two family members with an important renal phenotype and other typical manifestations of FD (white matter lesions and left ventricular hypertrophy) that only had the D313Y variant. These cases suggest that this variant of unknown significance may contribute to the development of common features of FD and should not be undervalued.
La enfermedad de Fabry (EF) es un trastorno de almacenamiento lisosómico multisistémico causado por mutaciones en el gen GLA que tienen como resultado una actividad deficiente o ausente de alfa-galactosidasa A. Existe un amplio espectro de variantes del gen GLA, algunas de las cuales se describen como no patógenas. La importancia clínica de la variante D313Y aún está en debate, aunque en los últimos años se ha considerado una variante de significado incierto o una variante benigna. A pesar de esta noción predominante, existen múltiples reportes de casos de pacientes con variante D313Y que presentaron signos y síntomas consistentes con EF sin ninguna otra explicación etiológica. En este artículo presentamos 2 familiares con un importante fenotipo renal y otras manifestaciones típicas de la EF (lesiones de la sustancia blanca e hipertrofia ventricular izquierda) que solo presentaban la variante D313Y. Estos casos indican que esta variante de significado incierto puede contribuir al desarrollo de características comunes de la EF y no debe subestimarse.
Fabry disease (FD) is an X-linked multisystem lysosomal storage disorder caused by mutations in the GLA gene. These mutations result in a deficient or absent activity of the lysosomal enzyme alpha-galactosidase A (αGal-A). This leads to a systemic accumulation of globotriaosylceramide (Gb3) and related glycosphingolipids in the plasma and cellular lysosomes of a wide range of organs and tissues, as podocytes and other cells in the kidneys, cardiomyocytes, vascular endothelial cells, cells of the nervous system and others.1
Gb3 deposition, however, may not be the only responsible for the organ manifestations, since it might exist other unexplained factors that could cause some features of FD, considering that disease manifestations may be present in the absence of severe deposits.2
The FD diagnosis is usually difficult since there are numerous possible manifestations and several different pathogenic variants with variable organ involvement. The mean delay to accurate diagnosis was estimated to be 13.7 years for males and 16.3 for females on the Fabry Outcome Survey.3
In the classic phenotype the first symptoms initiate during childhood such as neuropathic pain, renal manifestations (kidney injury and proteinuria), cutaneous lesions (angiokeratomas and telangiectasias), cornea verticillata and other unspecific symptoms (gastrointestinal disturbances, hypohidrosis, exercise intolerance). These symptoms are followed by transient ischaemic attacks and strokes, progressive renal insufficiency, concentric left ventricular hypertrophy, heart failure and typically premature death in the fourth or fifth decade of life.4
Heterozygous female patients have a vast range of possible manifestations and severity which are dependent on the X chromosome inactivation and the type of variant. Nevertheless, a large proportion of patients present with later-onset phenotypes with a great variety of age of onset, organ involvement and αGal-A levels.5
More than 900 variants in the GLA gene have been identified in the Human Gene Mutation Database.
There is still much debate about the pathogenicity of the D313Y variant, although in recent years it has been considered to be a benign variant or a variant of unknown significance (VUS), especially after the concept of LysoGb3 levels as the biomarker of FD. They are normally not increased in patients with D313Y variant. Nonetheless, there are several reports of patients with this variant that have important manifestations of FD with no other clinical or laboratory explanation. This will be covered in more detail in the discussion.
In this article we present 2 cases within the same family (mother and son) that carry the D313Y variant.
Case reportPatient 1An 18-year-old male presented to the emergency department with fatigue, hypertension, anorexia and weight loss. Laboratorial evaluation revealed severe anaemia (haemoglobin: 4.5g/dL) and kidney injury (creatinine: 18.9mg/dL, urea: 373mg/dL). Urinalysis was only positive for mild proteinuria (365mg/24h). Renal ultrasound showed small-sized kidneys with undifferentiated renal parenchyma. On the extensive laboratory analysis only C3 was mildly reduced (77mg/dL). Protein electrophoresis, immunoglobulins, free light chains, C4, antinuclear antibodies, ANCA, anti-GBM were within the normal reference ranges or were negative. Serologic profile of human immunodeficiency virus, hepatitis B and C were innocent. Echocardiogram displayed left ventricular hypertrophy (LVH). The cause of the kidney injury was not evident at the time of the clinical presentation. Kidney biopsy was not performed because of advanced kidney disease and kidney size. The patient was transplanted after six years on dialysis.
Patient 2A 43-year-old female was incidentally diagnosed with chronic kidney disease (CKD) on routine blood tests (creatinine: 2.9mg/dL, urea: 166mg/dL). By that time, the kidneys were small-sized and with undifferentiated renal parenchyma. Urinalysis did not show proteinuria or haematuria. The extensive laboratory analysis was inconclusive. Kidney biopsy was not performed for the same reason of the previous case. She started renal replacement therapy (RRT) after six years on follow-up. The patient presented with neurologic symptoms (neuropathic pain and decreased strength in the lower limbs, with a predominance of paresis in the right lower limb) one year before the CKD diagnosis. An MRI of the brain was preformed and revealed multiple, disseminated, hyperintense white matter lesions (WML) on the periventricular and subcortical areas (Fig. 1).
, Axial T2 FLAIR (B), Coronal T2 (C) – demonstrating disseminated punctuated and confluent WML on the periventricular and subcortical areas (patient 2).")
The patient 1, as mentioned in the introduction, is the son of the patient 2. The worsening of the neurologic symptoms of the patient 2 along with the kidney failure history of both patients raised the hypothesis of FD. Accordingly, the patient 1 was firstly tested and the αGal-A activity in dried blood spot (DBS) assay was partially decreased (2.84pmol per punch per h−1). Genetic testing revealed only the GLA variant D313Y (Exon 6, c.937G>T). Afterwards the patient 2 was tested and revealed the same variant, although with normal values of αGal-A activity. Lyso-Gb3 plasma levels were within the normal range in both patients.
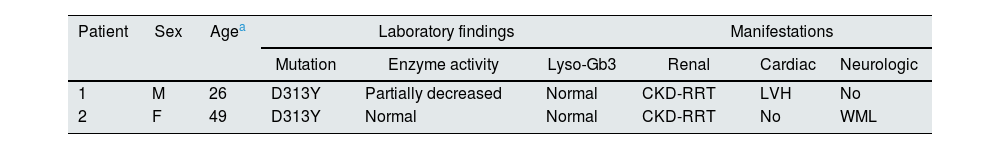
Both patients were evaluated for other symptoms of FD. Cornea verticillata, angiokeratomas, gastrointestinal symptoms (abdominal pain, vomiting, diarrhoea or constipation), hypohidrosis/hyperhidrosis were not detected. The renal, cardiac and neurological manifestations are represented in Table 1.
Summary of patient findings and manifestations.
| Patient | Sex | Agea | Laboratory findings | Manifestations | ||||
|---|---|---|---|---|---|---|---|---|
| Mutation | Enzyme activity | Lyso-Gb3 | Renal | Cardiac | Neurologic | |||
| 1 | M | 26 | D313Y | Partially decreased | Normal | CKD-RRT | LVH | No |
| 2 | F | 49 | D313Y | Normal | Normal | CKD-RRT | No | WML |
There are contradicting findings about the clinical importance of the D313Y variant in the literature. Yasuda et al.6 reported that in patients with this variant the expressed α-Gal A transported to the lysosomes had about 60% of the wild-type activity but resulted in a pseudodeficiency in plasma, due to its instability at neutral pH. This implies a lower D313Y mutant enzyme's activity in plasma than in leukocytes. They also suggest that the presence of the D313Y variant should prompt further investigation to detect a second causative mutation, since this can further impair the α-Gal A activity and/or stability and explain the disease manifestation in those patients. Oder et al.7 also corroborate that D313Y variant by itself does not lead to severe organ manifestations. They assessed 6 patients with that variant at baseline and during a 4-year follow-up. No significant manifestations were identified. Only one patient showed white matter lesions in the MRI but without neurological manifestations. Niemann et al.8 present 2 patients of the same family (father and daughter) with D313Y variant with reduced levels of α-Gal A activity, without relevant clinical manifestations of FD (the daughter had only unspecific pain in her arms) and LysoGb3 was found below the average in the daughter and undetectable in the father. This strengthens their assumption that this variant is not associated with FD with the current concept of lyso-Gb3 accumulation as a hallmark of the disease.9 Since there is significant activity of α-Gal A with the D313Y variant it only leads to a small amount of lyso-Gb3. Nevertheless, the pathogenic role of lyso-Gb3 is not well established since the mechanisms apart from lyso-Gb3 accumulation, that lead to organ manifestations, are still unknown.10,11
On the other hand, there is some clinical evidence in the literature that this variant may present with clinical features of FD. In some studies, it was identified a leading feature, more frequently cerebrovascular involvement followed by renal manifestations. Some cases presented with a later-onset phenotype. Yenicerioglu et al.12 tried to estimate the prevalence of FD in CKD patients not on dialysis and without a known cause (1453 CKD patients, with disclosure of 3 variants in mutation analysis) and found 2 D313Y carriers with moderate renal insufficiency (CKD stage G3b) and mild proteinuria. Data from another study13 that included 14 patients carrying the D313Y variant provided more evidence that this mutation might lead to organ manifestations compatible with FD: 4 patients had cerebrovascular involvement (stroke at a young age); 7 with acroparaesthesias/polyneuropathy; 2 presented cardiac hypertrophy but suffered from arterial hypertension; 1 had impaired renal function of unknown aetiology but without proteinuria; 1 with cornea verticillata. Another example of central nervous system involvement is in Lenders et al.14 also reported neurological involvement. They presented a family of 8 carriers of GLA D313Y and demonstrated the existence of multifocal WML in 7 carriers, without any other possible cause in the extensive investigation. None of those patients had other symptoms of FD. In another study,15 where they include 62 subjects demonstrating phenotypic traits suggestive of FD or belonging to families with a FD diagnosis, the authors identified 17 individuals with the D313Y variant. LysoGb3 was found normal in all patients. In the male subjects, except for one, had α-galA activity decreased in a range 56.2–87.5% compared to normal. In the group of D313Y variant, 4 were healthy but the rest presented characteristic manifestations of FD. Notably, 4 had end-stage renal disease (ESRD) on dialysis, 6 had WML, and also with other typical traits (acroparesthesias – 5, CKD not on dialysis – 2, LVH – 1, strokes of unknow origin – 1, cornea venticillata – 1, hearing loss – 1, GI symptoms – 1). Like in our case, one patient reported, in this article, initiated dialysis at a young age (25).
The manifestations of FD are nonspecific. Both our patients had unexplained renal insufficiency with mild or absent proteinuria, which is very atypical for the usual progression of the renal manifestations of FD. Normally, proteinuria is one of the initial kidney manifestations and it may initiate in the early teen years and more frequently during early adulthood. CKD progressively develops over time. The time of the presentation of end-stage renal failure in patient 1 is also highly unusual. Although, in an English cohort study of 98 hemizygous males, the mean age of ESRF was 37 years, the youngest ESFR patient presented at 18 years of age.16 One important limitation in our article was the unavailability of kidney biopsy, which we abdicated due to unacceptable risks without significant benefit. Patient 1 had only another trait of FD, LVH, but it may have other explanations like hypertension. As for patient 2, she has cerebral involvement with WML patent in the MRI and neurologic symptoms (paresis in the right lower limb and neuropathic pain in both lower limbs). Although these manifestations have some factors that do not fully fit in FD, no other explanation for them was identified despite thorough investigation.
ConclusionEven if D313Y variant is currently not considered to be pathogenic for presenting normal lyso-Gb3 values and for the majority of cases having a benign course, there is a considerable number of patients that presented with signs/symptoms consistent with FD without any other explanation. Although, at the same time, it is difficult to attribute them to FD as it has a great range of different presentations and there are important FD traits missing. Based in our findings in these particular patients, we cannot claim that their definite diagnosis is FD but this VUS should not be undervalued, and identified D313Y carriers may benefit of a consistent follow-up/assessment, expecting and monitoring possible manifestations of FD.
FundingThe authors received no financial support for the research, authorship, and/or publication of this article.
Conflict of interestAll the authors declared no competing interests.



