












Presentamos un paciente con una glomeruloesclerosis focal y segmentaria en el que los estudios genéticos evidenciaron la coexistencia de 2 mutaciones simultáneas, una en el gen que codifica la podocina y otra en el gen que codifica el colágeno tipo IV. Se describe la presentación clínica, que fue atípica, el procedimiento de elaboración diagnóstica y la estrategia terapéutica, así como la evolución clínica del paciente, la pobre respuesta terapéutica y las implicaciones de esta entidad en el trasplante renal.
INTRODUCCIÓN
La glomeruloesclerosis focal y segmentaria (GSFS) es una lesión histológica que se caracteriza por la presencia de esclerosis de un segmento del glomérulo y que afecta focalmente a algunos glomérulos. Su incidencia es variable, entre el 13-20% de las biopsias renales, según edad y área geográfica1. Como aspectos de interés en su etiopatogenia, pueden ser de causa primaria por alteración podocitaria asociada a la presencia de factores circulantes, secundaria a mecanismos adaptativos (p. ej., reflujo, hipertensión arterial) o debida a factores genéticos, por mutaciones en los genes que codifican proteínas de la barrera de filtración glomerular. Todas estas circunstancias dan lugar a proteinuria, que puede ser nefrótica, y en un porcentaje importante de casos, si esta no se controla, progresar hacia la enfermedad renal crónica (ERC).
La GSFS de causa genética en el 25-40% de los casos familiares se asocia a mutaciones en el gen que codifica la podocina (NPHS2). Sin embargo, en los últimos años se han descrito nuevas mutaciones que afectan a genes que codifican otras proteínas de la barrera de filtración glomerular o incluso varias mutaciones simultáneas en distintos genes, algunos de los cuales se asociaban clásicamente con otras patologías distintas de la GSFS. Estas circunstancias han obligado a modificar la aproximación y las estrategias de actuación ante un paciente que presente lesiones de GSFS, en el que se debe intentar determinar la etiología y el posible origen genético de este hallazgo.
CASO CLÍNICO
Varón de 16 años, que acude a urgencias por presentar un cuadro catarral de 1 semana de evolución que se acompaña de astenia, fiebre y visión borrosa. En los antecedentes personales destaca en la infancia la presencia de microhematuria y proteinuria aislada, con seguimiento ocasional e irregular por pediatría. Seis meses antes del presente cuadro presentó episodio de sinusitis maxilar, que no recibió tratamiento. En los antecedentes familiares (fig. 1) refería padre y 2 tíos paternos con microhematuria. También 2 familiares del abuelo materno fallecieron en diálisis.
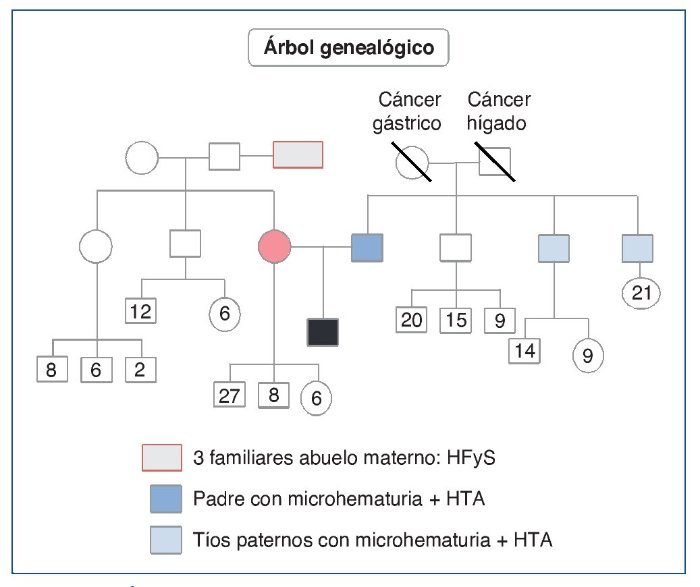
Figura 1. Árbol genealógico.
HFyS: hialinosis focal y segmentaria; HTA: hipertensión arterial.
Exploración física: presión arterial (PA), 160/110 mmHg; 39 ºC; fondo de ojos con retinopatía hipertensiva grado III, y edemas maleolares. Resto dentro de los límites normales.
Como datos analíticos más relevantes en urgencias destacaban en la bioquímica: Cr, 5,8 mg/dl; urea, 151 mg/dl; K, 5,7 mEq/l; P, 5,3 mEq/l; albúmina, 2,3 g/l, lactato deshidrogenasa (LDH), 264 UI/l. En el hemograma: Hb (hemoglobina), 10,4 g/dl; linfocitos 2.810/μl; reticulocitos, 28%, y plaquetas, 158.000/μl. En sedimento: proteinas ++++ y 30-50 hematíes/campo. En los estudios analíticos posteriores se observó: colesterol, 257 mg/dl; PTH (hormona paratiroidea), 112 UI/l, y en orina: proteinuria, 10,8 g/día.
El resto de las pruebas complementarias: hormonas tiroideas, normales; Coombs D e I, negativos; estudio inmunológico y complemento, normales. Proteinograma, normal y ausencia de cadenas ligeras libres. Serologías y cultivos, negativos; ASLO (anticuerpos antiestreptolisina O), normal. Ecografía: riñones hiperecogénicos (RD, 10 cm/RI, 9,8 cm). Angio-TC renal, normal. Audiometría normal.
Con el diagnóstico de síndrome nefrítico, se realizó biopsia renal percutánea. La muestra incluía 34 glomérulos por sección, 25 de ellos (73%) globalmente esclerosados con restos de material hialino. De los 7 glomérulos restantes, 2 no mostraban alteraciones relevantes (fig. 2), mientras que los demás presentaban intensa proliferación endocapilar segmentaria o, más frecuentemente, global con ocasional cariorrexis y marcada hiperplasia/ hipertrofia del epitelio visceral (podocitos), con formación de seudosemilunas (figs. 3 y 4). No se identificó colapso de los capilares glomerulares ni lesiones en el polo tubular de glomérulos. Los podocitos hiperplásicos mostraban signos de reabsorción proteica y, además, se apreciaban áreas de adherencia flóculo-capsular en 2 de los glomérulos (fig. 5). También se apreciaban amplios parches de atrofia tubular, con intenso infiltrado inflamatorio acompañante y fibrosis intersticial, por lo general coincidiendo con glomérulos esclerosados, que suponen aproximadamente un 40% del área cortical. De forma ocasional se observaban túbulos dilatados con contenido proteico y/o hemático. No se identificaron depósitos hialinos en las paredes arteriolares, pero todas ellas mostraron una llamativa hipertrofia concéntrica de la capa muscular con marcada reducción y casi obliteración de la luz capilar (fig. 6). Las arterias interlobulares/ arcuatas mostraban menor grado de hiperplasia que la media sin alteraciones significativas de la íntima. No se observaron focos de necrosis fibrinoide ni trombos capilares en los glomérulos ni en las arteriolas. En el estudio de inmunofluorescencia directa se apreciaban únicamente depósitos glomerulares irregulares de IgM (1+) y C3 (2+) (fig. 7). Se realizó estudio de microscopia electrónica a partir del material incluido en parafina, y los hallazgos fueron de valoración limitada debido a la mala preservación tisular. Seis de los 7 glomérulos de la muestra se encontraban globalmente esclerosados y el restante mostraba esclerosis segmentaria; en su zona no esclerosada se observó pérdida de integridad de los endotelios y pérdida parcial de integridad de las células epiteliales viscerales, con intensa fusión de los pedicelos en las áreas mejor conservadas (fig. 8). La falta de integridad de las paredes capilares no permitía una medición correcta de su grosor, aunque, en las láminas densas conservadas se apreciaban algunas irregularidades y algunas laminaciones, así como zonas de menor grosor y zonas de mayor severidad en el espesor de la membrana de la lámina densa. No se observaron depósitos electrón-densos. Con estos hallazgos se diagnosticó de glomeruloesclerosis focal y segmentaria, variante celular.
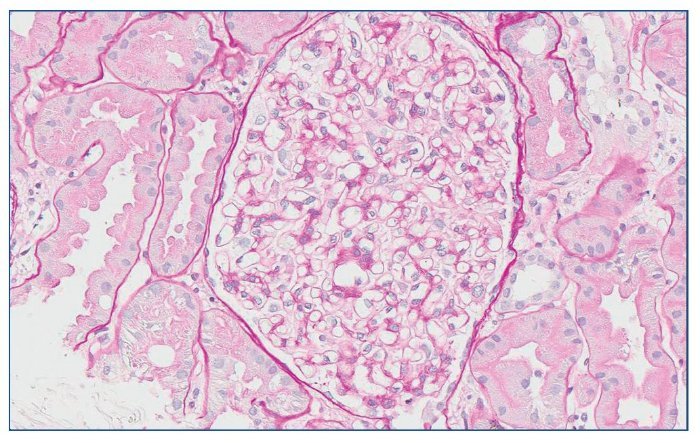
Figura 2. Glomérulo sin alteraciones relevantes. PAS ×20.
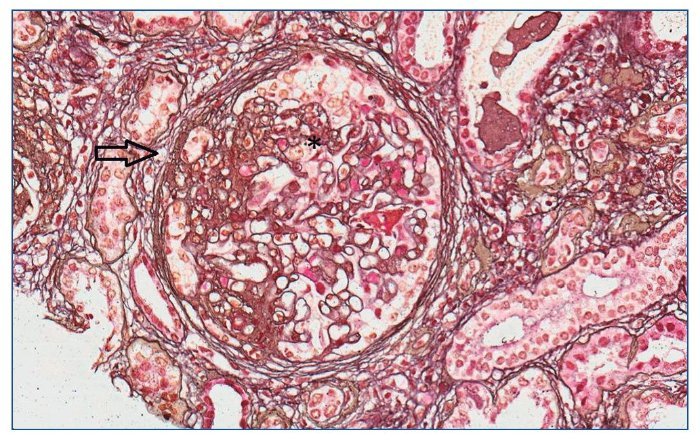
Figura 3. Lesión esclerosante que afecta aproximadamente al 40% del ovillo glomerular (flecha) con hipercelularidad endocapilar segmentaria (*). Plata de Jones ×20.
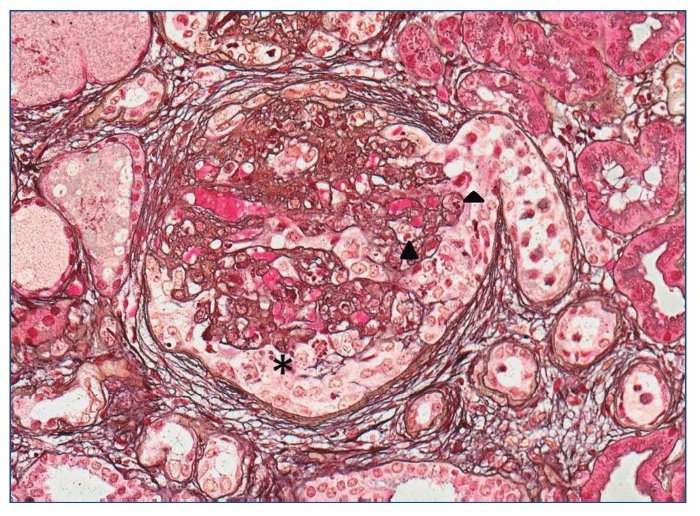
Figura 4. Proliferación endocapilar global (puntas de flecha) que oblitera las luces capilares. Llamativa hipertrofia podocitaria (*) con formación de seudosemilunas. Ocasionales figuras de mitosis. Plata de Jones ×20.
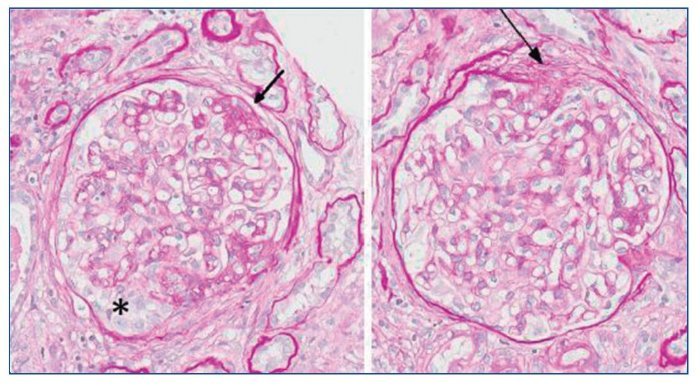
Figura 5. Lesiones esclerosantes segmentarias con adherencia a la cápsula de Bowman (flecha) y área de hipertrofia podocitaria con formación de seudosemiluna (*). PAS ×20.
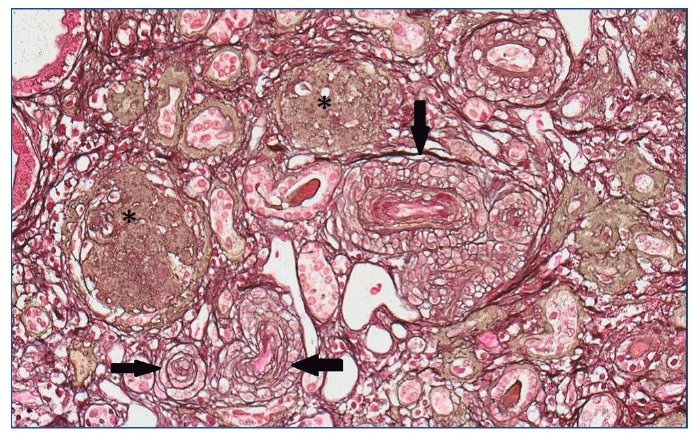
Figura 6. Glomérulos globalmente esclerosados (*). Arteriolas con marcada proliferación miointimal concéntrica con obliteración casi completa de las luces vasculares (flechas).
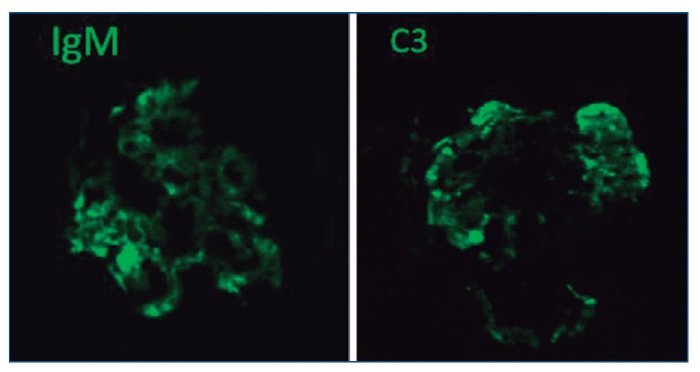
Figura 7. Depósitos glomerulares granulares, toscos, focales y segmentarios, de IgM y C3. Inmunofluorescencia directa.
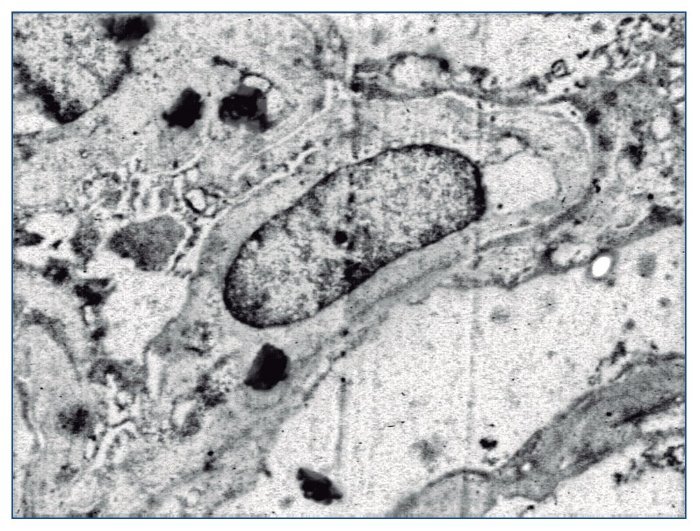
Figura 8. Fusión de pedicelos podocitarios. ME ×2.800.
Ante la forma de presentación y las sospechas diagnósticas iniciales, se comenzó tratamiento mediante bolos de 6-metilprednisolona (3 dosis de 500 mg), para continuar con prednisona oral y, dado el deterioro de la función renal, se inició hemodiálisis. En los días posteriores, tras los hallazgos histológicos y en espera de los estudios genéticos, se asoció tratamiento con micofenolato mofetilo (500 mg/12 h). Tras 4 meses sin respuesta y ausencia de recuperación de la función renal, se retiraron el micofenolato y la prednisona. Se transfirió para tratamiento sustitutivo renal mediante diálisis peritoneal automatizada (DPA).
El estudio genético mostró: ausencia de mutaciones en genes del complemento; se objetivaron 3 variantes en heterocigosis, 2 mutaciones en el gen NPHS2 (R229Q y A284V) (asociadas a GNFS autosómica recesiva), además de una mutación en el gen COL4A4 (asociada al síndrome de Alport y hematuria familiar) (figs. 9 a 11).
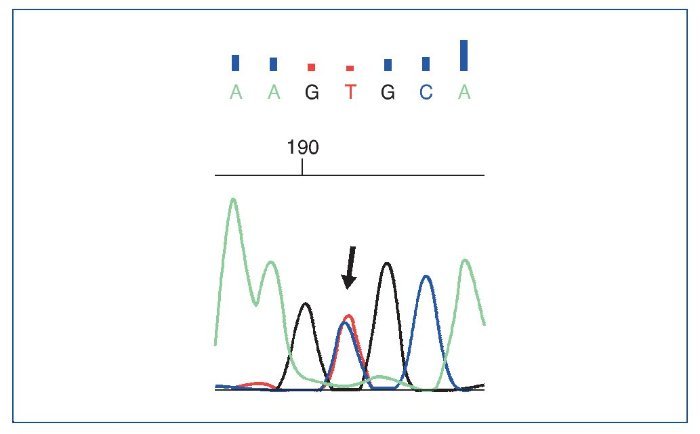
Figura 9. Estudio genético. Cambio c.851C>T encontrado en el gen NPHS2.
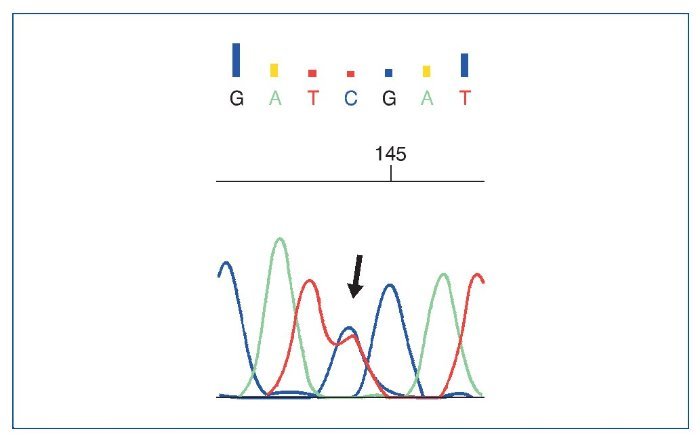
Figura 10. Cambio c.686G>A (C>T en su forma reversa) encontrado en el gen NPHS2.
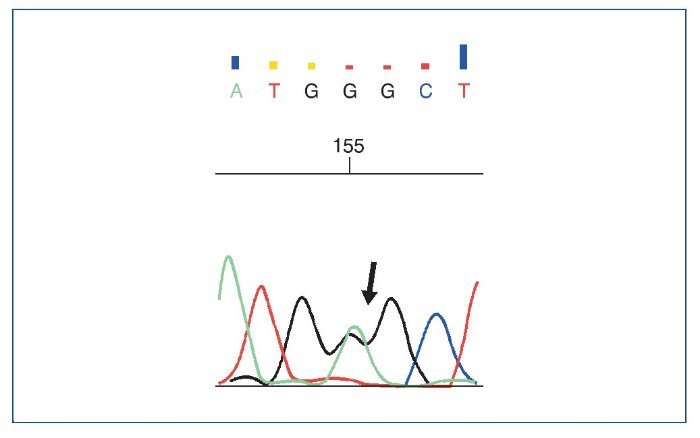
Figura 11. Cambio c.2690G>A encontrado en el gen COL4A4.
El paciente se incluyó en lista de espera de trasplante renal; se desestimó la donación de vivo emparentado por si pudieran ser portadores de dichas mutaciones. Tras 24 meses en DPA recibió trasplante de donante en asistolia (Maastricht III), con muy buena compatibilidad. Recibió inmunosupresión de inducción con timoglobulina, esteroides, tacrolimus y micofenolato mofetilo, con función renal retrasada y posterior mejoría progresiva de la función del injerto. Al cuarto día postrasplante presentó un hematoma retroperitoneal y, dado que había presentado varios episodios de hemorragia durante los procedimientos invasivos (p. ej., biopsia renal, catéter peritoneal), se realizó estudio de coagulación, que detectó un déficit parcial de factor de von Willebrand. En el momento actual (16 meses postrasplante) presenta una evolución favorable, con función renal estable (Cr, 1,6 mg/dl), con buen estado clínico y adecuado control de la PA.
DISCUSIÓN
Nos encontramos ante un paciente adolescente que debuta con un síndrome nefrítico, en el que se observó la presencia de una GSFS y que los estudios genéticos permitieron determinar la coexistencia de mutaciones correspondientes a esta entidad junto a hallazgos de un síndrome de Alport. En la aproximación clínica inicial hay que tener en cuenta que la causa más frecuente de síndrome nefrítico tras un cuadro infeccioso en niños o jóvenes es la presencia de una glomerulonefritis aguda, generalmente posinfecciosa. Cabe destacar que nuestro paciente, en el momento del debut, presentaba un daño renal grave que era bien tolerado, y con síntomas posiblemente asociados a la presencia de hipertensión grave, lo que orientaba hacia un posible proceso evolucionado a lo largo de un tiempo prolongado. En este sentido, destacaba que el paciente tenía antecedentes en la infancia de episodios aislados de alteraciones en el sedimento (microhematuria, 5-10 hematíes/campo, y proteinuria ++ en tira reactiva), por lo que se valoró en alguna ocasión sin un seguimiento regular posterior. Esta circunstancia nos hizo plantear la existencia de una posible glomerulonefritis de base y que en el momento actual estábamos ante la evolución final de una ERC. A esta posibilidad se añadían los hallazgos ecográficos, en los que ambos riñones mostraban un patrón con áreas corticales hiperecogénicas. Sin embargo, la sospecha clínica no concordaba con la exploración física, en la que no había estigmas claros de ERC (p. ej., buen estado nutricional, adecuado desarrollo estaturoponderal), lo que hacía probable la presencia de un proceso intercurrente sobreañadido como causa del empeoramiento y precipitación del cuadro (cabe recordar que el paciente debutó en el seno de un síndrome febril y una hipertensión arterial [HTA] grave) y se tratara de un fracaso renal agudo, cuya etiología estaría por determinar. Estas posibilidades diagnósticas y la necesidad de iniciar un tratamiento hicieron imprescindible la realización de una biopsia renal. Como ya se ha descrito, el diagnóstico histológico fue de una GSFS que, de manera más característica, presentaba una afectación arteriolar muy grave. A partir de este hallazgo, el otro posible diagnóstico que era obligado descartar era el de una microangiopatía trombótica asociada a HTA maligna (cifras, 160/110 mmg, y retinopatía hipertensiva grado III) o bien a un síndrome hemolítico urémico atípico. La ausencia de un patrón hematológico y bioquímico compatible con esta entidad (plaquetas, haptoglobina y LDH normales, Coombs directo negativo, frotis de sangre periférica normal) permitía descartarlos de manera razonable. Por otro lado, los resultados de los marcadores inmunológicos no orientaban a una enfermedad autoinmune como causa del cuadro clínico o de los eventos que lo precipitaron.
Respecto al tratamiento que recibió nuestro paciente, como primera opción previa a la biopsia renal se administraron pulsos de metilprednisolona, además de tratamiento antibiótico de amplio espectro y fármacos antihipertensivos, y se inició hemodiálisis. Una vez se obtuvo el resultado histológico, a la espera de los estudios genéticos, se planteó la posibilidad de tratamiento inmunosupresor; la primera opción fueron los inhibidores de la calcineurina, que se desestimaron dado el importante deterioro de la función renal; lo mismo ocurrió con otras opciones (p. ej., ciclofosfamida), dados la escasa posibilidad de respuesta, los efectos secundarios y la edad del paciente. Como alternativa, a la espera de disponer de todos los resultados, se optó por mantener una pauta de prednisona (1 mg/kg en descenso progresivo) y micofenolato mofetilo (500 mg/12 h), que se suspendieron tras 4 meses debido a la ausencia de respuesta y continuó dependiente de diálisis.
El estudio genético mostró 3 variantes génicas patológicas en heterocigosis, 2 mutaciones en el gen NPHS2 (R229Q y A284V) (figs. 9 y 10), asociadas a GSFS autosómica recesiva, además de una mutación en el gen COL4A4 (fig. 11), asociada al síndrome de Alport y hematuria familiar. En relación con estos hallazgos, se ha descrito que la combinación de 2 mutaciones de NPHS2, siendo una de ellas la variante R229Q, se relaciona con la GSFS de aparición en la edad pediátrica tardía o adulta (especialmente en Europa occidental)2, que corresponde a lo que se encontró en nuestro paciente. A pesar de que el alelo R229Q se ha considerado un polimorfismo poblacional (el 3% de la población europea y en la de Sudamérica con ascendencia hispana), se ha descrito su patogenicidad para desarrollar GSFS. La variante R229Q (en heterocigosis), en combinación con otra mutación patogénica de NPHS2, se asocia al síndrome nefrótico cortico-resistente con edad de presentación y desarrollo de ERC terminal de forma más tardía que pacientes con 2 mutaciones patogénicas3; este podría ser el motivo por el que nuestro paciente debutara a una edad más tardía siendo portador de mutaciones del gen de la podocina. Además, la mutación A284V en combinación con R229Q (en heterocigosis) se ha descrito frecuentemente en pacientes de Chile y Argentina, y posteriormente en población española4.
En relación con la tercera mutación encontrada en el gen COL4A4, esta se asocia al síndrome de Alport con herencia de predominio autosómica recesiva, pero también dominante5 y asociado a hematuria familiar, principalmente autosómica dominante6. Sin embargo, también se han observado mutaciones en genes del colágeno tipo IV en un porcentaje importante de pacientes con GSFS, de hasta un 38% en las formas familiares y en un 3% en las formas esporádicas (en los genes COL4A3, COL4A4 y COL4A5)7. Por este motivo, en pacientes que presentan GSFS es necesario extender el estudio genético a los genes del colágeno tipo IV. Como dato relevante, se han observado casos de enfermedad glomerular justificados por interacción génica entre distintas mutaciones8.
Debemos destacar que la prevalencia de las formas monogénicas de GSFS, especialmente en adultos, está infraestimada, debido a que presentan un fenotipo muy variable. Además, identificar las causas genéticas es complejo dado que en ocasiones no hay evidencia de antecedentes familiares conocidos y que existen mutaciones con penetrancia incompleta. Por este motivo, varios autores recomiendan estudiar un posible origen genético en las GSFS en el adulto que presenta antecedentes familiares de esta, en GSFS primarias resistentes al tratamiento inmunosupresor y en las GSFS secundarias sin causa evidente9. Nuestro paciente tenía varios familiares por rama paterna con microhematuria y 3 familiares por rama materna habían tenido GSFS. Dados los hallazgos en el estudio genético del paciente, se les realizó estudió genético a la madre, que mostró que era portadora de la variante génica A284V en heterocigosis, y al padre, que era portador de la variante génica R229Q en homocigosis y de la variante G897E en heterocigosis (fig. 9). Como dato adicional, el paciente presenta un déficit leve del factor de von Willebrand. No hemos encontrado relación entre la GSFS, la enfermedad de Alport y el déficit de factor de von Willebrand.
Por tanto, podemos concluir que la detección de un origen genético de la GSFS es de suma importancia, especialmente para evitar terapias inmunosupresoras ineficaces. Además, el estudio genético tiene implicaciones en el trasplante, pues la tasa de recidiva en la GSFS primaria está en torno al 30%10, mientras que en las de origen genético raramente ocurre (2,5%)11. Sin embargo, podría tener implicaciones en el trasplante de donante vivo emparentado, dado que sería posible que la enfermedad se desarrollara en el injerto12 o podría desarrollarla el donante en evolución, por este motivo se descartó esta opción en nuestro paciente. Tras 24 meses en tratamiento con DPA, recibió un injerto de donante fallecido y en la actualidad presenta un buen estado clínico, con una aceptable función renal.
La GSFS continúa siendo un reto para el nefrólogo, no solo en el momento del diagnóstico, sino también al decidir el tratamiento, en el posible riesgo de recidiva en el injerto y en la detección de nuevas mutaciones genéticas asociadas.
Conflicto de intereses
Los autores declaran que no existe conflicto de intereses.
Correspondencia:
Patricia García García
Servicio de Nefrología.
Hospital Nuestra Señora de la Candelaria.
Carretera del Rosario, 145. 38010 Santa Cruz de Tenerife.
patri102@hotmail.com
Revisión por expertos bajo la responsabilidad de la Sociedad Española de Nefrología.


