Consulte aquí la exposición del caso
RESOLUCIÓN DEL CASO
AMILOIDOSIS SECUNDARIA (TIPO AA) COMO CAUSA DE DETERIORO DE LA FUNCIÓN RENAL
Autores: Álvarez Muñoz A.S.1, Ramos Tomás M.C.1, Garrigós Almerich E.V.1, López Romero L.C.1 Moreno Abenza G.2, Pérez Rojas J.2, Peris Domingo A.1, Hernandez Jaras J.1
1 Servicio de Nefrología y 2 Servicio de Anatomía Patológica del Hospital Universitari i Politècnic La Fe. Valencia (Valencia).
Correspondencia: angie_asam24@hotmail.com
INTRODUCCIÓN
Amiloidosis son un grupo de enfermedades que se caracterizan por el depósito en los tejidos de diferentes órganos de un material amorfo, que presenta una estructura fibrilar característica en la microscopía electrónica y un patrón de láminas beta-plegadas. [1].
Son enfermedades poco frecuentes, la incidencia estimada se ha calculado en algunos estudios realizados en diversos países: uno de ellos de largo seguimiento concluyó que la incidencia de amiloidosis sistémica es aproximadamente 0.8 por 100 000 habitantes. La incidencia de la amiloidosis AL ajustada a edad, se estimó a partir de uno de los estudios más destacados sobre la epidemiología de esta patología, con una tasa de 5,1-12,8 casos por millón persona-años. Sin embargo, al existir casos detectados por autopsia, sugiere que la incidencia podría ser mayor. [2,3,4].
Se pueden clasificar según la extensión de los depósitos en: sistémicas y localizadas, así como también por el tipo de proteína precursora fibrilar causante. Existen 18 tipos de amiloidosis sistémicas y 22 de localizadas. Las más frecuentes son: amiloidosis sistémica AL, amiloidosis por transtiretina y amiloidosis secundaria AA. [1].
El riñón es uno de los órganos más frecuentemente afectado en las formas sistémicas siendo fundamental para su diagnóstico la realización de una biopsia renal.
La amiloidosis AA también llamada amiloidosis reactiva está asociada a procesos inflamatorios crónicos: desordenes del tejido conectivo, infecciones crónicas, enfermedad intestinal inflamatoria, síndrome paraneoplásico, alteraciones genéticas asociadas como por ejemplo la fiebre mediterránea familiar. El amiloide precursor es el amiloide A sérico (SAA), una apolipoproteína de alta densidad producida por los hepatocitos bajo la regulación de citosinas proinflamatorias incluyendo a: interleucina 1 (IL-1), interleucina 6 (IL-6), interferón G y factor de necrosis tumoral (TNFa); su concentración sérica está asociada con la actividad inflamatoria. [5].
CASO CLÍNICO
Paciente de 51 años que es remitido desde atención primera a consultas externas de Nefrología por deterioro de la función renal y proteinuria. A destacar los siguientes antecedentes personales:
- Hipertensión arterial de 9 años de evolución en tratamiento con Manidipino con buen control
- Dislipemia de 9 años de evolución en tratamiento Simvastatina.
- Episodios de infecciones urinarias (3 episodios en un solo año) secundarias a incontinencia urinaria de esfuerzo que fue resuelta mediante tratamiento quirúrgico no volviendo a presentar nuevos episodios de infección.
- Cuadro de intertrigo candidiásico inguinal crónico con gran extensión en pared abdominal
- Eccema crónico pruriginoso y severo confirmado mediante biopsia cutánea y con buena respuesta a tratamiento con corticoides.
- Rinitis alérgica crónica con pruebas cutáneas positivas para polen y controlada con con Ebastina.
- Quiste de retención maxilar izquierdo como hallazgo casual en TC dental, valorado por otorrinolaringología y corroborado por TC facial, que no precisó ningún tratamiento. (ver Figura 1)
Figura 1: Quiste de retención en suelo de seno maxilar izquierdo. Mínimo engrosamiento mucoso de suelo de seno maxilar derecho. Resto de senos paranasales libres.
-Sin antecedentes familiares de patología renal.
En la primera visita en consultas externas de nefrología se confirma los hallazgos analíticos compatibles con enfermedad renal crónica estadio IIIa con creatinina de 1,25 mg/dl, FG de 45 ml/min, iones en rango de normalidad, metabolismo fosfocálcico con PTH de 59,00 pg/mL, Vit D 28.3 ng/ml, calcio de 9,2 mg/dL, fosforo de 4,3 mg/dL, no presentaba anemia ni déficit de hierro. Sedimento urinario sin hematuria ni leucocituria y en la orina de 24 horas presentaba proteinuria de 0,55 g/24h. En la ecografía renal, los riñones eran de tamaño normal, con buena diferenciación cortico medular e hipertrofia de columna de Bertin en el riñón izquierdo, sin dilatación de vía urinaria.
Durante el seguimiento posterior en consultas externas, la paciente presenta deterioro progresivo de la función renal: creatinina 2.70 mg/dl, proteinuria en rango subnefrótico con niveles de albumina normal y sedimento de orina inactivo. El resto del estudio, la autoinmunidad era normal, niveles de complemento en rango de normalidad, serología vírica negativa, proteinograma normal con ausencia de banda monoclonal en suero y en orina. La exploración física sin hallazgos significativos, no edema en miembros inferiores y buen control de las cifras tensionales con un solo fármaco.
Dados los antecedentes infecciosos, inflamatorios e infecciosos que presentaba, se decide ampliar el estudio obteniendo niveles elevados de IgE > 5.000 kUA/L y proteína amiloide sérica A de 19,70 mg/L (rango normal de 0,00 - 6,99).
Se decide realizar biopsia renal: (ver figura 2)
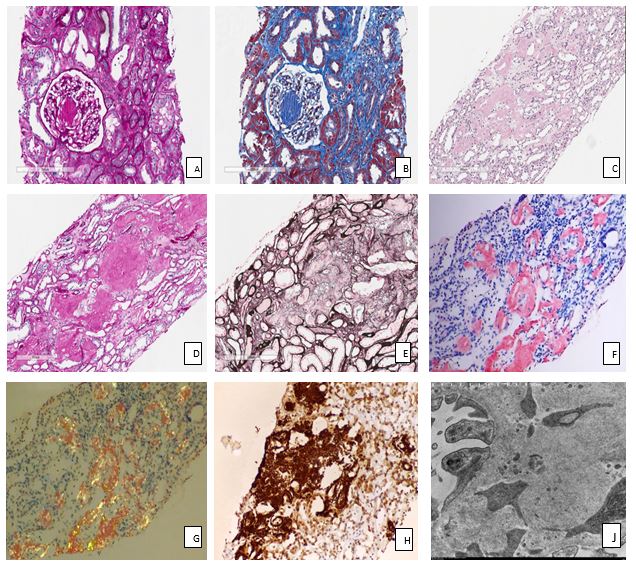
Fig. 2: Imágenes del estudio histológico y ultraestructural de la biopsia renal
A: tinción de PAS, con el objetivo de 20X, en la que observamos depósitos de material PAS positivo a nivel de los glomérulos.
B: tricrómico de Masson, a 20X, en la que se observan depósitos de material acelular a nivel de los glomérulos, que se tiñe de coloración azulada mediante esta tinción.
C: hematoxilina-eosina, a 10X del intersticio renal. Se observan depósitos de material eosinofílico acelular distribuidos de manera intersticial entre los túbulos colectores medulares.
D: tinción de PAS, a 15X. Se observan ausencia de tinción de los depósitos anteriormente descritos.
E: tinción de Jones, a 15X. Se identifica ausencia de tinción del material acelular estudiado.
F: tinción de Rojo Congo, a 15X. Se observa tinción rojo congófila en los depósitos acelulares intersticiales.
G: luz polarizada del Rojo Congo, en la que se observa positividad verde manzana de los depósitos congófilos.
H: microscopia electrónica, en la que se identifican fibrillas intersticiales de entre 7 y 13 nm a nivel de la membrana basal glomerular.
Diagnóstico histológico: Nefropatía por amiloidosis AA: fibras intersticiales de 7-13 mm positivas con tinción Rojo Congo y con inmunohistoquímica para amiloide tipo AA. Patrón de lesión: glomeruloesclerosis mesangial difusa y arteriolar. Moderados signos de lesión tubular aguda, leves signos de atrofia y fibrosis intersticial.
Debido a que la paciente presentaba niveles elevados de IgE, episodio de eccema, candidiasis y hallazgo de quiste de retención maxilar; se decide ampliar estudio genético para mutaciones relacionadas con Síndrome Hiper IgE: exones y regiones intrónicas flanqueantes de los genes DOCK8, STAT3, IKBKG, IL21R y SPINK5, grandes deleciones y duplicaciones del gen DOCK8 y STAT3; los resultados fueron negativos.
El síndrome de hiperIgE, inicialmente llamado Síndrome de Job, se caracteriza clínicamente por dermatitis e infecciones recurrentes, aunque existe una significante variación de signos y síntomas interindividual. La clínica incluye: abcesos cutáneos (74%), eccema 58%, otras patologías alérgicas (alergia a medicamentos 43%, alergia a alimentos 38%, alergia ambiental 18%), retención de quistes dentarios 41%, fracturas 39%, escoliosis 34%, cáncer 7%. Los pacientes casi siempre tienen niveles séricos elevados de IgE, generalmente en un rango desde 1000 hasta más de 50000 unidades/Ml. [6].
El diagnóstico de Síndrome de hiperIgE se basa en los hallazgos clínicos y de laboratorio, con la confirmación diagnóstica del estudio molecular en la que se identifica una mutación como por ejemplo mutación en el gen STAT3.
La mayoría de pacientes con Síndrome IgE AD tienen un defecto genético en la vía de señalización y activación de la transcripción 3 (STAT3), la cual está codificada en el cromosoma 17q21 (MIM #147060). Variantes patogénicas que causan desórdenes HiperIgE-like han sido identificadas hasta ahora en ocho genes, como por el ejemplo: gen tirosina quinasa 2 (TYK2), gen de la citoquinesis (DOCK8), gen de transductor de señalización de interleucina 6 (IL6ST), deficiencia del receptor de interleucina 6 (IL6R), entre otros. Los defectos de STAT3 producen disfunción de los linfocitos Th17. En los pacientes que no tienen variantes patogénicas detectables de STAT3 también se puede encontrar diferenciación y función defectuosa de linfocitos Th17. [7,8].
Los pacientes con síndrome hiperIgE o trastornos HiperIgE-like que no tienen un defecto genético identificado podrían tener defectos en los elementos reguladores de STAT3, DOCK8, TYK2 o un defecto genético nuevo más allá de las vías de señalización. [7,8].
DISCUSIÓN
La incidencia de amiloidosis estimada en la clínica es baja, pero en realidad es una patología infradiagnosticada que produce afectación renal en un porcentaje considerable: 70% de los casos de amiloidosis AL y más del 95% en amiloidosis AA.
Las manifestaciones clínicas de la amiloidosis son muy variadas, dependen del subtipo de proteína, del patrón y de la severidad de depósito en cada órgano afectado. El diagnóstico muchas veces se retrasa por la inespecificidad y variabilidad de la clínica. [9].
Las manifestaciones a nivel renal están relacionadas con la localización: glomerular, intersticial o vascular, y el grado de infiltración amiloidea. Es frecuente la proteinuria, síndrome nefrótico y la evolución a enfermedad renal crónica. [10].
-Si existen depósitos a nivel glomerular suele expresarse como proteinuria en rango nefrótico, sedimento urinario típicamente inactivo, creatinina normal o moderamente elevada.
-Si existen depósitos a nivel vascular y tubular es común el desarrollo de enfermedad renal crónica lentamente progresiva con mínima o nula proteinuria.
Otras manifestaciones son:
-Nefropatía por depósitos de cilindros de amiloide intratubulares: muy poco frecuente, se presenta como fracaso renal agudo.
-Glomerulonefritis con semilunas: Muy rara, casi todos los casos están reportados en pacientes con amiloidosis AA debido a artritis reumatoide o sus variantes.
Los factores pronósticos para el desarrollo de enfermedad renal crónica son: el grado disfunción renal en el momento del diagnóstico y el grado de proteinuria. Una vez desarrollada, la supervivencia se reduce. Por lo dicho previamente destaca la importancia de considerar esta patología como diagnóstico diferencial del deterioro de la función renal. [11].
El diagnóstico definitivo se realiza mediante la biopsia renal, demostrando la presencia de depósitos de amiloide: infiltrados Rojo Congo positivo, mediante inmunohistoquímica y/o técnicas de proteómica.
En nuestra paciente, la presencia de proteinuria con sedimento inactivo, el estudio inmunológico de patología glomerular fue negativo, incluidos IEF de plasma y orina que descartó gammapatia monoclonal, junto al deterioro de la función renal y los antecedentes infecciosos, inflamatorios y alérgicos con aumento de proteína C reactiva y proteína amiloide A, nos llevó a realizar la biopsia renal que confirmó el diagnóstico de Amiloidosis AA.
En el tratamiento de amiloidosis AA, el control de la patología causante subyacente inflamatoria, infecciosa o tumoral es fundamental para el reducir o suprimir la producción de la proteína amiloide A. Por ejemplo, la Colchicina es el tratamiento estándar en caso de amiloidosis secundaria a fiebre mediterránea familiar (FMF); agentes activos contra citosinas proinflamatorias (IL-1 beta, TNF alf, IL-6) han demostrado eficacia en algunos casos de amiloidosis AA secundaria a patologías reumáticas y a enfermedades hereditarias autoinflamatorias. Por ejemplo, Anakinra (anti IL-1) se usa para paciente con FMF que no han respondido a Colchicina.
Tocilizumab, es un anticuerpo monoclonal recombinante que inhibe la actividad biológica en mediada por IL-6 en las células. Varios reportes de casos (en algunos están incluidos pacientes con Amilodosis AA de causa no filiada), un estudio retrospectivo comparativo, y el primer estudio nacional de pacientes con Amiloidosis AA en Japón han mostrado que Tocilizumab disminuye los niveles de la proteína amiloide A, mejora los síntomas y produce una regresión del depósito la proteína amiloide. [12,14].
Okuda et al. compararon la eficacia del tratamiento con anti-TNF-a y Tocilizumab en 42 pacientes con Artritis Reumatoide con amiloidosis AA. Con Tocilizumab la media de niveles de proteína AA fue menor después del tratamiento, el filtrado glomerular aumentó en 73% de los pacientes y la remisión o menor actividad de artritis reumatoide fue más frecuente. [13].
Por otra parte, el Eprosidato, un mimético de glucosaminoglicano que fue desarrollado para interferir con la formación de Amiloidosis AA, fue retirado tras la fase 3 de estudio por no mostrar beneficio.
Para nuestra paciente se planteó inicio de tratamiento con Tocilizumab (anti-IL6), tras realizar radiografía de tórax, quantiferón, ecocardiograma con resultados son normales, se actualizó la serología vírica y se remitió a medicina preventiva para vacunación. El tratamiento ha sido bien tolerado, con mejoría evidente de parámetros de inflamación: normalizando proteína C reactiva y descenso de proteína sérica amiloide A; además mejoría de función renal con creatinina 2.21 mg/dl FG 25 ml/min, y reducción de proteinuria 0,47 gr, manteniendo sedimento de orina inactivo.
CONCLUSIONES
El diagnóstico etiológico del deterioro de la función renal implica un reto significativo, especialmente cuando nos enfrentamos a patologías con clínica tanto inespecífica como variada, y sobretodo poco comunes como la amiloidosis. Esta enfermedad puede implicar un pronóstico desfavorable a nivel de función renal, así como a nivel de supervivencia del paciente; precisamente por esa razón hay que destacar la importancia de considerar su existencia y probabilidad de que esté presente en el paciente que tenemos frente a nosotros, y así instaurar el tratamiento respectivo.
Tocilizumab es un anticuerpo monoclonal anti IL-6 capaz de suprimir los niveles de proteína amiloide AA, producir una regresión de los depósitos de esta proteína y generar mejoría clínica de los síntomas de amiloidosis secundaria; convirtiéndose en una estrategia terapéutica importante de esta patología.
BIBLIOGRAFÍA Y REFERENCIAS
1.- Benson MD, B. J. (2018). Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Epub 2019 Jan 7, Dec;25(4):215-219.
2.- Pinney JH, Smith CJ, Taube JB, Lachmann HJ, Venner CP, Gibbs SD, Dungu J, Banypersad SM, Wechalekar AD, Whelan CJ, Hawkins PN, Gillmore JD. Systemic amyloidosis in England: an epidemiological study. Br J Haematol. 2013 May;161(4):525-32. doi: 10.1111/bjh.12286. Epub 2013 Mar 11. PMID: 23480608; PMCID: PMC4296340.
3.- Robert A. Kyle, Athena Linos, C. Mary Beard, Reinhold P. Linke, Morie A. Gertz, W. Michael O'Fallon, Leonard T. Kurland, Incidence and Natural History of Primary Systemic Amyloidosis in Olmsted County, Minnesota, 1950 Through 1989,Blood,Volume 79, Issue 7,1992,Pages 1817-1822, ISSN 0006-4971,https://doi.org/10.1182/blood.V79.7.1817.1817. (https://www.sciencedirect.com/science/article/pii/S0006497120736785)
4.- Panizo N, Rivera F, López-Gómez JM; Spanish Registry of Glomerulonephritis. Decreasing incidence of AA amyloidosis in Spain. Eur J Clin Invest. 2013 Aug;43(8):767-73. doi: 10.1111/eci.12097. Epub 2013 May 18. PMID: 23683125.
5.-Papa R, Lachmann HJ. Secondary, AA, Amyloidosis. Rheum Dis Clin North Am. 2018 Nov;44(4):585-603. doi: 10.1016/j.rdc.2018.06.004. Epub 2018 Sep 7. PMID: 30274625.
6.- Gernez Y, Freeman AF, Holland SM, Garabedian E, Patel NC, Puck JM, Sullivan KE, Akhter J, Secord E, Chen K, Buckley R, Haddad E, Ochs HD, Fuleihan R, Routes J, Muskat M, Lugar P, Mancini J, Cunningham-Rundles C. Autosomal Dominant Hyper-IgE Syndrome in the USIDNET Registry. J Allergy Clin Immunol Pract. 2018 May-Jun;6(3):996-1001. doi: 10.1016/j.jaip.2017.06.041. Epub 2017 Sep 19. PMID: 28939137; PMCID: PMC5858974.
7.-Woellner C, Gertz EM, Schäffer AA, Lagos M, Perro M, Glocker EO, Pietrogrande MC, Cossu F, Franco JL, Matamoros N, Pietrucha B, Heropolitanska-Pliszka E, Yeganeh M, Moin M, Español T, Ehl S, Gennery AR, Abinun M, Breborowicz A, Niehues T, Kilic SS, Junker A, Turvey SE, Plebani A, Sánchez B, Garty BZ, Pignata C, Cancrini C, Litzman J, Sanal O, Baumann U, Bacchetta R, Hsu AP, Davis JN, Hammarström L, Davies EG, Eren E, Arkwright PD, Moilanen JS, Viemann D, Khan S, Maródi L, Cant AJ, Freeman AF, Puck JM, Holland SM, Grimbacher B. Mutations in STAT3 and diagnostic guidelines for hyper-IgE syndrome. J Allergy Clin Immunol. 2010 Feb;125(2):424-432.e8. doi: 10.1016/j.jaci.2009.10.059. PMID: 20159255; PMCID: PMC2878129.
8.-Kumánovics A, Wittwer CT, Pryor RJ, Augustine NH, Leppert MF, Carey JC, Ochs HD, Wedgwood RJ, Faville RJ Jr, Quie PG, Hill HR. Rapid molecular analysis of the STAT3 gene in Job syndrome of hyper-IgE and recurrent infectious diseases. J Mol Diagn. 2010 Mar;12(2):213-9. doi: 10.2353/jmoldx.2010.090080. Epub 2010 Jan 21. PMID: 20093388; PMCID: PMC2871728.
9.-D'Aguanno, V., Ralli, M., Artico, M. et al. Systemic Amyloidosis: a Contemporary Overview. Clinic Rev Allerg Immunol 59, 304-322 (2020). https://doi.org/10.1007/s12016-019-08759-4
10.-Gupta N, Kaur H, Wajid S. Renal amyloidosis: an update on diagnosis and pathogenesis. Protoplasma. 2020 Sep;257(5):1259-1276. doi: 10.1007/s00709-020-01513-0. Epub 2020 May 24. PMID: 32447467
11.-Uda H, Yokota A, Kobayashi K, Miyake T, Fushimi H, Maeda A, Saiki O. Two distinct clinical courses of renal involvement in rheumatoid patients with AA amyloidosis. J Rheumatol. 2006 Aug;33(8):1482-7. PMID: 16881107.
12.-Courties A, Grateau G, Philippe P, Flipo RM, Astudillo L, Aubry-Rozier B, Fabreguet I, Fahd W, Fain O, Guggenbuhl P, Hachulla E, Papo T, Richez C, Sibilia J, Morel J, Berenbaum F, Sellam J; Club Rhumatismes Inflammation and the REGATE Registry. AA amyloidosis treated with tocilizumab: case series and updated literature review. Amyloid. 2015;22(2):84-92. doi: 10.3109/13506129.2014.1002031. Epub 2015 Jan 14. PMID: 25585627.
13.-Okuda Y. AA amyloidosis - Benefits and prospects of IL-6 inhibitors. Mod Rheumatol. 2019 Mar;29(2):268-274. doi: 10.1080/14397595.2018.1515145. Epub 2018 Oct 2. PMID: 30132351.
14.-Yasuaki Okuda, AA amyloidosis - Benefits and prospects of IL-6 inhibitors, Modern Rheumatology, Volume 29, Issue 2, 4 March 2019, Pages 268-274, https://doi.org/10.1080/14397595.2018.1515145





