




Focal segmental glomerulosclerosis (FSGS) is classified into three forms: primary, secondary, and genetic FSGS. Genetic FSGS is defined as sporadic or familial types. The mutations in the gene inverted formin (INF)2 are mostly encountered in familial genetic FSGS cases. A 29-year-old female patient without any parental consanguinity and family history of kidney disease, who had nephrotic syndrome with inactive urine sedim and normal glomerular filtration rate was diagnosed as kidney biopsy-proven FSGS. She had partial remission under treatment of prednisone and cyclosporine. The patient was re-evaluated due to presence of relapse in proteinuria during her pregnancy. Genetic analysis revealed a heterozygous missense variant (NM_022489.4:c.653G>A; p.R218Q) in the INF2 gene. This case report presents a young female patient with sporadic FSGS induced by INF2 mutation.
La glomeruloesclerosis focal y segmentaria (GEFS) se clasifica en tres formas: primaria, secundaria y genética. La GEFS genética se define como esporádica o familiar. Las mutaciones en el gen INF2 se encuentran principalmente en casos de GEFS genética familiar. Una paciente de 29 años, sin consanguinidad parental ni antecedentes familiares de enfermedad renal, con síndrome nefrótico con sedimentación urinaria inactiva y tasa de filtración glomerular normal, fue diagnosticada con GEFS confirmada mediante biopsia renal. Presentó remisión parcial bajo tratamiento con prednisona y ciclosporina. La paciente fue reevaluada debido a la presencia de una recaída de proteinuria durante el embarazo. El análisis genético reveló una variante heterocigótica sin sentido (NM_022489.4: c.653G>A; p.R218Q) en el gen INF2. Este informe de caso presenta el caso de una paciente joven con GEFS esporádica inducida por la mutación INF2.
Focal segmental glomerulosclerosis (FSGS), defined as presence of characteristic light microscopic (LM) findings in kidney biopsy (segmental glomerular hyalinosis and/or sclerosis without specific immune deposits) is responsible for nearly 30% of the cases with nephrotic syndrome. The pathogenesis and treatment of FSGS differ between the patients based on the classification which includes primary, secondary, and genetic.1
The prevalence of genetic FSGS (sporadic or familial) can change from less than 5–30% of all FSGS cases.2 Familial FSGS are nearly 1% of all FSGS cases.3 At the initial time of diagnosis, absence of nephrotic syndrome, presence of immunosuppressive drug-resistant nephrotic syndrome, family history for FSGS, and syndromic presentation in a patient whom diagnosed as FSGS on kidney biopsy require genetic screening.2,4 The most common genetic defects responsible for FSGS development in adults change based on the type of genetic mutation. For sporadic cases, mutations in the genes for nephrin (NPHS1) were commonly encountered, however, for monogenic cases, mutations in the gene inverted formin (INF)2 were mostly detected.2,5
Here, we present the outcome and follow up of a patient whom diagnosed as genetic FSGS secondary to sporadic mutation in INF2 gene four years ago.
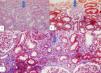
Case presentationA 29-year-old female patient without any parental consanguinity and family history of kidney disease, who had previously been diagnosed as hypothyrodism and nephrotic syndrome (proteinuria of 4.7g/d) with inactive urine sedim and estimated glomerular filtration rate (eGFR) of 137mL/min/1.73m2 (by CKD-EPI based formula) secondary to kidney biopsy-proven FSGS during her first pregnancy when she delivered a preterm (36 week) and low birth weight (2000g) infant four years ago, and prescribed levothyroxine (50mcg/d), prednisone (0.5mg/kg/d), calcium+vitamin D, and cyclosporine (2.5mg/kg/d) was re-evaluated due to presence of relapse in proteinuria (Fig. 1). Kidney biopsy which had been performed four years ago, had revealed global glomerular sclerosis in 5 out of 26 glomeruli, segmental glomerular sclerosis in 1 out of 26 glomeruli, mild–moderate cortical interstitial fibrosis (25%), subintimal arterial hyalinosis, by light microscopy (LM) (Fig. 2). However, fibrinoid necrosis, crescents, and diffuse basal membrane thickenings were not detected. There was no immune deposition (C3, C1q, C4d, Ig G, Ig A, Ig M, amyloid A) by immunofluorescence microscopy (IFM). Autoantibodies (such as antinuclear, anti-dsDNA, anti-Ro-52, anti-Ro/SSA, anti-centromere, anti-La/SSB, anti-Scl 70, anti-Smith, anti-glomerular basement membrane, anti-cardiolipin and anti-neutrophil cytoplasmic antibodies), and hypocomplementemia (C3 and C4 levels) were not detected.


After clinical evaluation of the patient and absence of complete remission despite the optimum treatment Schedule, we performed Common Inherited Disorders Panel, which includes 3324 genes [nephrin (NPHS1), podocin (NPHS2), alpha-actinin-4 (ACTN4), transient receptor potential cation channel subfamily C member 6 (TRPC6), phospholipase C epsilon 1 (PLCE1), etc.]. Genetic analysis revealed a heterozygous missense variant (NM_022489.4:c.653G>A; p.R218Q) in the INF2 gene. This alteration is located in exon 4 of the INF2 gene and results in the substitution of an arginine (R) at position 218 with a glutamine (Q). According to the guidelines of the American College of Medical Genetics and Genomics (ACMG), this variant is classified as pathogenic.6
The patient had been treated with previous drugs and followed with proteinuria of 0.7–1.49g/day, serum creatinine of 0.75–0.8mg/dL, and eGFR of 103–114mL/min/1.73m2. Unexpectedly, she had become pregnant again. Although the treatment regimen continued, during second pregnancy, proteinuria increased from 0.73g/d to 4.76g/d, serum albumin levels decreased from 4.18g/dL to 3.0g/dL, eGFR mildly increased to 116–120mL/min/1.73m2. During breastfeeding, she did not take cyclosporine, hence, proteinuria increased to 7.5g/d and serum creatinine levels increased from 0.69mg/dL to 0.94mg/dL. After reusage of cyclosporine, proteinuria decreased to 1.41g/d but serum creatinine levels increased to 1.1mg/dL (Fig. 1).
DiscussionHuman formin proteins (inverted formin 1 and 2) were first reported in 2008 as the major regulatory proteins for structural and functional integrity of actin filament and other cytoskeletal proteins which also maintain podocyte plasticity.7 The first research identifying the association of INF2 gene (located on chromosome 14) defects with familial autosomal dominant (AD) FSGS [with or without Charcot-Marie-Tooth disease (CMT), identified by demyelinating peripheral neuropathy] appearance was published in 2010.8 Since then, genetic screening of FSGS patients resulted that these mutations have been among the most common mutations of the genetic FSGS.3 Brown et al. reported the presence of heterozygous INF2 gene mutations in 12% of families with FSGS.8 Similarly, Gbadegesin et al. found that mutations in INF2 gene were responsible for the 16.3% of the AD FSGS cases.3 However, no INF2 gene mutations were reported to be detected in individuals with idiopathic (sporadic) disease or those with autosomal recessive (AR) FSGS in their study.3 The most commonly mutated gene which has been reported to be found in sporadic FSGS patients is NPHS1.5
The genetic location of the FSGS caused by INF2 mutations has been found on 14q32.33 (https://omim.org/entry/610982).8 The locus of exon 4 of this gene has been reported to contain nearly most of the FSGS-causing variants.3 The variant in the INF2 gene of the present case has been previously reported and is not novel. Although there is a link between FSGS induced by INF2 gene mutations and CMT, peripheral neuropathy which is the main manifestation of CMT, was not found in our patient. The INF2 mutations are more commonly encountered in patients with both FSGS and CMT (75%) than FSGS solo (12–17%).9 In addition, INF2 gene mutations have been very rarely reported in sporadic FSGS patients.10 Barua et al. searched the presence of INF2 mutations among 281 individuals with sporadic FSGS and they have found only 2 cases. Based on this research, they reported that INF2 mutations were rarely found in sporadic cases of FSGS.
INF2-mutated familial FSGS patients have variable clinical penetrance with the first presentation and onset of kidney failure approximately at the ages of 20–40 and 30–40.10 Sporadic cases with INF2 mutations are very rare, as far as we know, less than 5 cases are reported in the literature. Therefore, the clinical perspective of these cases is not clear, childhood onset has been documented in these rare cases.10,11 Finally the accurate treatment for these sporadic cases is not present.
In conclusion, unplanned pregnancies of patients with proteinuria lead to exacerbation of nephrotic syndrome and preclampsia as maternal outcome, preterm births (defined as <37 weeks) and low birth weight (defined as 2500g) as fetal outcome.12 Exacerbation of maternal proteinuria and preterm delivery also occurred in our case. Our case is important because it presents the presence, clinical presentation, renal outcome during 4-year of follow up, and achievement of partial remission with prednisone and cyclosporine in a young female patient with sporadic FSGS induced by INF2 mutation.
ORCID IDİbrahim Erbay: 0000-0003-0004-2449
Hakan Ertan: 0009-0003-8624-2376
Beyhan Güvercin: 0000-0003-4157-4513
Sevdegül Aydın Mungan: 0000-0002-4882-352X
Neslihan Cinkara: 0000-0003-4869-0891
CRediT authorship contribution statementKubra Kaynar: Conception, design, analysis and interpretation of data, drafting the article and revising it. İbrahim Erbay: Documentation of the patient data, literature research. Hakan Ertan: Literature research and interpretation of data. Beyhan Güvercin: Literature research. Sevdegül Aydın Mungan: Interpretation of pathological findings. Neslihan Cinkara: Interpretation of genetic analysis.
Compliance with ethical standardsWritten informed consent regarding this article was obtained from the patient.
Informed consentWritten informed consent was obtained from the patient regarding the publishment of this manuscript.
Declaration of generative AI and AI-assisted technologies in the writing processNo AI was used for this manuscript.
FundingNo fund was taken.
Conflicts of interestThe authors declare that they have no conflicts of interest.



