
X-linked hypophosphatemia (XLH) has an incidence of 1/20,000 newborns.1 It is caused by loss-of-function mutations of the PHEX gene, resulting in excess circulating fibroblast growth factor 23 (FGF23), with renal loss of phosphate and decreased synthesis of 1,25-dihydroxyvitamin D.2 Chronic hypophosphatemia leads to rickets and osteomalacia, growth retardation, lower limb malformations, pain and decreased physical function, mobility and quality of life.1,3 Conventional replacement therapy (phosphate salts and 1,25-dihydroxyvitamin D4 does not correct hypophosphatemia and may induce hypercalciuria, nephrocalcinosis and hyperparathyroidism. Moreover, it is difficult to implement, especially in children, given the gastrointestinal side effects and the need for frequent dosing.3–5 Burosumab (a recombinant human monoclonal antibody against FGF23) improves renal tubular phosphate reabsorption (RTP) with increased levels of phosphorus and 1,25-dihydroxyvitamin D.6
We conducted an observational, longitudinal, retrospective study at a referral hospital in Bahía Blanca, Argentina, to report the long-term outcome of patients with genetically confirmed XLH rickets from 1997 to 2022. Demographic and clinical data at diagnosis, imaging studies, and initial and subsequent laboratory results were recorded. Healthcare workers conducted check-ups every 3–6 months and a dental examination annually. A jugal mucosa sample was collected from one member of each family for a next-generation sequencing panel of 13 genes with different inheritance patterns. Treatment-related data were also obtained.
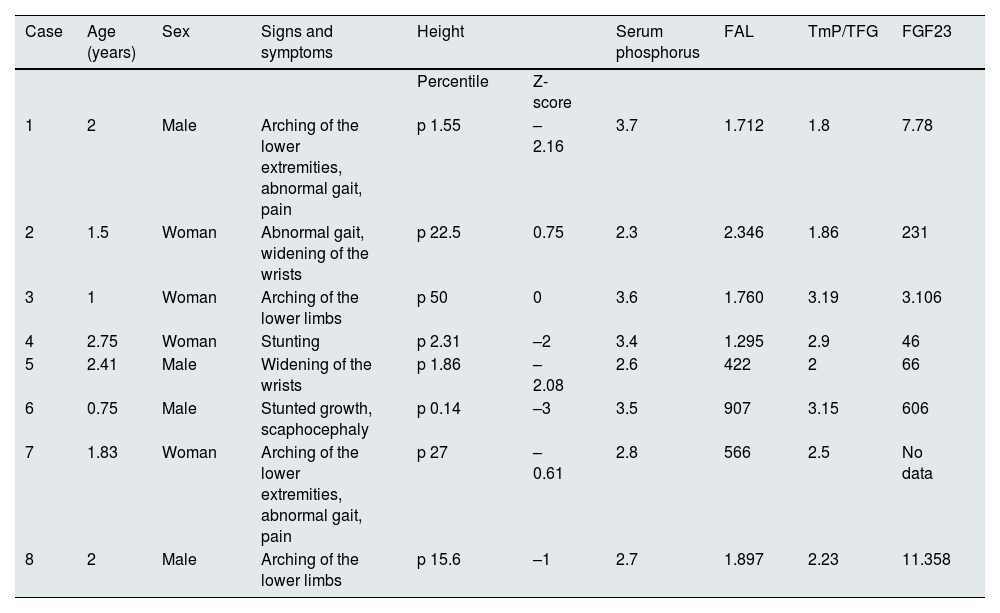
Eight patients (four males) with a median age at diagnosis of 1.9 years (range: 0.75–2.75), followed for a median of 7.16 years (range: 2–24), were included. Six patients were members of two families; the other two patients had de novo variants that had not been previously described (Table 1). Clinical manifestations and laboratory data at diagnosis are shown in Table 2. Half were growth retarded; disproportionate short stature, lower limb malformations and abnormal gait were observed in most patients. All had limitations in daily activities (jumping, running, walking), severe radiological abnormalities (cupped metaphysis, widened and irregular epiphyses of long bones), low phosphorus levels, normal calcium and parathormone values, and high alkaline phosphatase levels. Four patients had RTP < 85% and all participants were characterized by maximal tubular reabsorption of phosphate per glomerular filtration rate (TmP/GFR) <3.8 mg/dl. FGF23 levels were variable (Table 2).
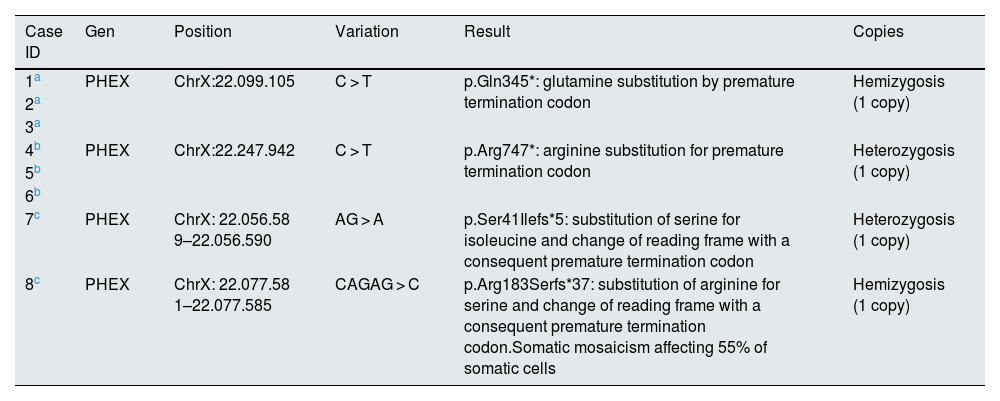
PHEX gene mutation.
| Case ID | Gen | Position | Variation | Result | Copies |
|---|---|---|---|---|---|
| 1a | PHEX | ChrX:22.099.105 | C > T | p.Gln345*: glutamine substitution by premature termination codon | Hemizygosis (1 copy) |
| 2a | |||||
| 3a | |||||
| 4b | PHEX | ChrX:22.247.942 | C > T | p.Arg747*: arginine substitution for premature termination codon | Heterozygosis (1 copy) |
| 5b | |||||
| 6b | |||||
| 7c | PHEX | ChrX: 22.056.58 9–22.056.590 | AG > A | p.Ser41Ilefs*5: substitution of serine for isoleucine and change of reading frame with a consequent premature termination codon | Heterozygosis (1 copy) |
| 8c | PHEX | ChrX: 22.077.58 1–22.077.585 | CAGAG > C | p.Arg183Serfs*37: substitution of arginine for serine and change of reading frame with a consequent premature termination codon.Somatic mosaicism affecting 55% of somatic cells | Hemizygosis (1 copy) |
First family: the initial patient (case 1) was genetically confirmed. He had four siblings, one of them (case 2) and a daughter (case 3) with the same diagnosis.
Clinical and biochemical findings at diagnosis.
| Case | Age (years) | Sex | Signs and symptoms | Height | Serum phosphorus | FAL | TmP/TFG | FGF23 | |
|---|---|---|---|---|---|---|---|---|---|
| Percentile | Z-score | ||||||||
| 1 | 2 | Male | Arching of the lower extremities, abnormal gait, pain | p 1.55 | –2.16 | 3.7 | 1.712 | 1.8 | 7.78 |
| 2 | 1.5 | Woman | Abnormal gait, widening of the wrists | p 22.5 | 0.75 | 2.3 | 2.346 | 1.86 | 231 |
| 3 | 1 | Woman | Arching of the lower limbs | p 50 | 0 | 3.6 | 1.760 | 3.19 | 3.106 |
| 4 | 2.75 | Woman | Stunting | p 2.31 | –2 | 3.4 | 1.295 | 2.9 | 46 |
| 5 | 2.41 | Male | Widening of the wrists | p 1.86 | –2.08 | 2.6 | 422 | 2 | 66 |
| 6 | 0.75 | Male | Stunted growth, scaphocephaly | p 0.14 | –3 | 3.5 | 907 | 3.15 | 606 |
| 7 | 1.83 | Woman | Arching of the lower extremities, abnormal gait, pain | p 27 | –0.61 | 2.8 | 566 | 2.5 | No data |
| 8 | 2 | Male | Arching of the lower limbs | p 15.6 | –1 | 2.7 | 1.897 | 2.23 | 11.358 |
ALP: alkaline phosphatase; FGF23: fibroblast growth factor 23; TmP/GFR: maximal tubular phosphate reabsorption per glomerular filtration rate.
Reference values (1–3 years of age): phosphorus = 3.9−6 mg/dl; FAL = 116–294 UI/l; TmP/TFG = 3.8−5 mg/dl; FGF23 = 0–134 pg/ml.
Cases 1 to 3 had the same mutation.
Cases 4 to 6 had the same mutation.
Cases 7 and 8 had de novo mutations.
After diagnosis, all patients received continuous phosphate replacement therapy with 5 daily doses of phosphate salts (average: 40 mg/kg/day) and 0.25 μ g/day of 1,25-dihydroxy vitamin D. Adherence was difficult due to gastrointestinal intolerance; improvement of radiological rickets lesions was observed in the first year of treatment, without restoration of phosphorus levels. In the long term, all patients had persistent radiological signs of rickets and residual bone malformations. Four patients maintained short stature. No patient developed hypercalciuria, nephrocalcinosis, hyperparathyroidism or dental abscesses.
Due to persistent rickets, musculoskeletal pain, limitation of daily activities and fractures, two children and two adults were rotated to subcutaneous burosumab after receiving replacement therapy for a median of 6 and 18 years, respectively. Children received 0.8 mg/kg every two weeks; adults, 1 mg/kg every four weeks, with discontinuation of conventional therapy two weeks earlier. After starting burosumab, all patients reported significant improvement in physical activities, with less fatigue and no pain. Height gain was observed in two patients who started burosumab during infancy, reaching a Z-score of– 0.2. Normalization of phosphorus and TmP/GFR was verified. Radiological signs of rickets disappeared in all these patients. No serious adverse reactions were observed.
In patients with XLH, phosphate supplementation can stimulate FGF23 levels and renal phosphate excretion, with a vicious cycle that limits its efficacy.3 Conventional therapy is unsuccessful in many cases, and up to two-thirds of children require lower limb surgery.7 Burosumab has been shown to be safe and effective, with improvement in TmP/GFR, hypophosphatemia, linear growth and functional capacity, as well as decrease in pain and severity of rickets.6,8,9 Four of our patients were rotated to burosumab, with clinical and biochemical improvement and complete resolution of radiological signs of rickets. These results were most notable in children.
We conclude that, in our long-term study with continuous follow-up, the main finding is the marked improvement of biochemical, radiological and especially clinical manifestations in patients who rotated from conventional treatment to burosumab.
Conflict of interestThe authors report no conflicts of interest.



