


Atypical haemolytic uraemic syndrome is a disease caused by complement regulation abnormalities that generally progresses to chronic end-stage renal disease with a high rate of recurrence in kidney transplantation and a high risk of graft loss. Anti-complement therapy has improved the prognosis of these patients, achieving disease remission in most cases, increasing the likelihood of a successful kidney transplant and increasing patient and graft survival. Drugs with low risk of induction of thrombotic microangiopathies such as belatacept and mycophenolate have also been used with satisfactory results. We present the case of a young patient at high immunological risk, with atypical haemolytic uraemic syndrome due to factor H mutation, who underwent a successful kidney transplantation with eculizumab, thymoglobulin, belatacept, mycophenolate and steroids, to date preserving excellent graft function without disease recurrence.
El síndrome hemolítico urémico atípico es una enfermedad relacionada con alteración en la regulación del complemento que generalmente evoluciona a enfermedad renal crónica terminal, con alta tasa de recaída en el trasplante renal y elevado riesgo de pérdida del injerto. La terapia anticomplemento ha mejorado el pronóstico de estos pacientes, logrando tener remisión de la enfermedad en la mayoría de los casos, aumentando la posibilidad de un trasplante renal exitoso e incrementando la supervivencia del paciente y del injerto; igualmente el uso de medicamentos con bajo riesgo de inducción de microangiopatías trombóticas como el belatacept y micofenolato se han utilizado con resultados satisfactorios. Presentamos el caso de una paciente joven de alto riesgo inmunológico, con síndrome hemolítico urémico atípico por mutación del factor H, a quien se realizó trasplante renal exitoso con eculizumab, timoglobulina, belatacept, micofenolato y esteroides conservando excelente función del injerto y sin recaídas de su enfermedad.
Thrombotic microangiopathies (TMAs) are a group of conditions characterised by the presence of non-immune microangiopathic haemolytic anaemia, thrombocytopaenia and single or multiple organ failure. There are multiple causes associated with the onset of TMA; they may be secondary to a deficit in ADAMTS13 enzyme activity, known as thrombotic thrombocytopaenic purpura. It may also be seen after enteroinvasive infections due to Shiga toxin-producing bacteria, giving rise to typical haemolytic uraemic syndrome. Other TMAs are secondary to complement-amplifying conditions such as pregnancy, infections, autoimmune diseases, neoplasms, drugs, or toxins; or due to changes in alternative complement pathway regulation which causes atypical haemolytic uraemic syndrome (aHUS), also known as thrombotic microangiopathy, secondary to changes in complement regulation.1,2 Prior to 2011, when anticomplement therapy was not yet available, the latter condition was associated with a high risk of terminal chronic kidney disease and death3; and, if these patients received a kidney transplant, graft survival was very low, due to a high percentage of recurrence of aHUS post transplant. For this reason, a simultaneous liver-kidney transplant was one of the options available for these patients. However, this procedure had a high risk of complications and death.3
Since the appearance of eculizumab, it has been possible to perform kidney transplants in patients with aHUS, with better short- and medium-term outcomes; decreasing recurrence, improving kidney graft survival and even reducing the possibility of rejection mediated by antibodies.4
Below we present the case of a patient with a diagnosis of terminal chronic kidney disease secondary to aHUS and sensitised due to multiple transfusions, who received a successful kidney transplant with a protocol of eculizumab, thymoglobulin, and belatacept.
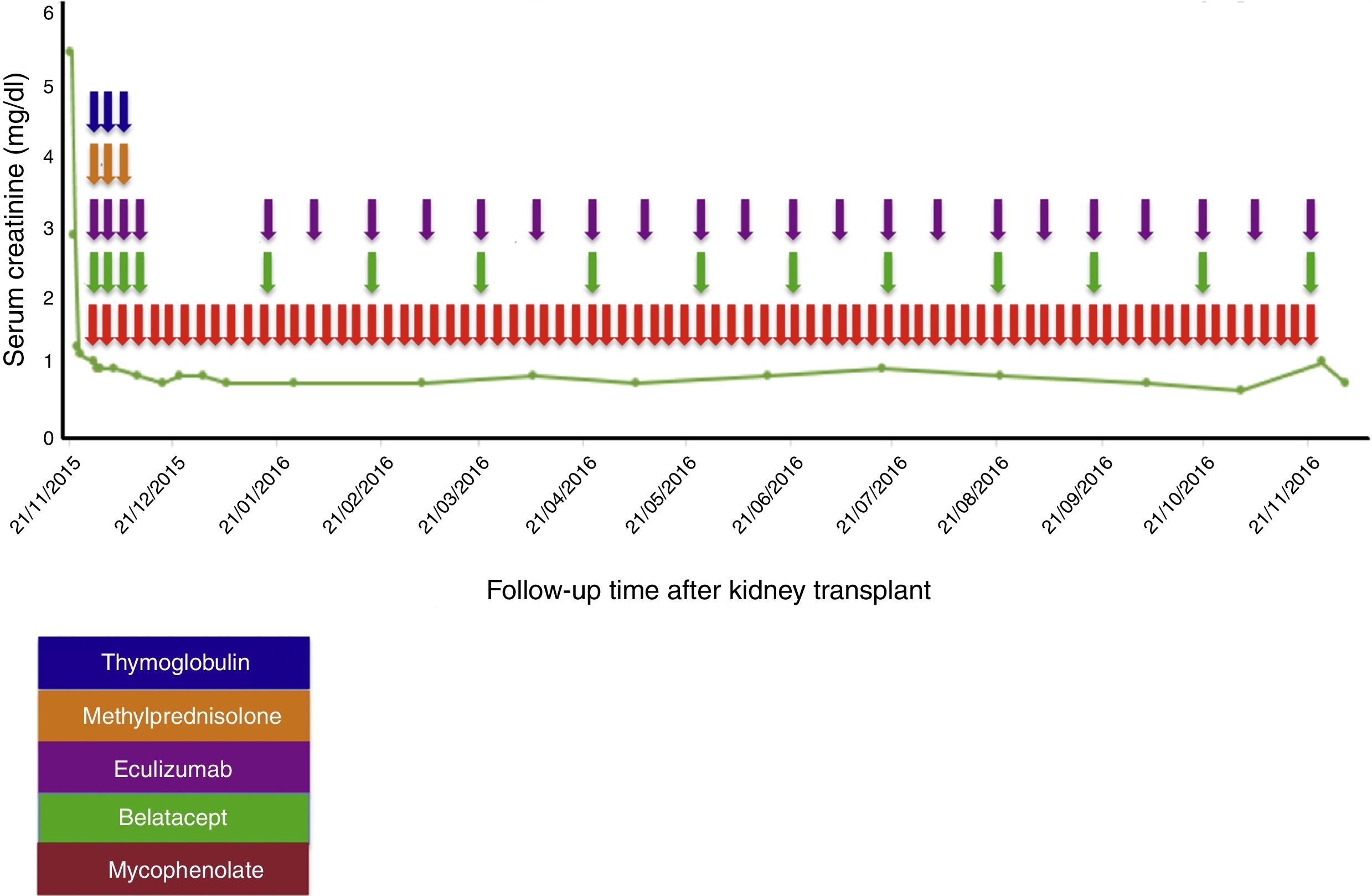
Case reportA 38-year-old female patient, with a history of TMA diagnosed in 2005, in whom infectious, immunological, toxic, metabolic and neoplastic causes were ruled out. At the time of diagnosis, the patient received treatment with plasma exchange (30 sessions), multiple transfusions (20U of red blood cells), steroids, intravenous (IV) cyclophosphamide (six 750mg doses at one-month intervals) and IV rituximab (four 500mg doses at one-week intervals); therapy to which she was refractory; with progressive deterioration of kidney function until reaching terminal chronic kidney disease and a need for haemodialysis-type renal replacement therapy since 2005. In 2008, she was put on the kidney transplant waiting list at another institution. At that time, she presented with a class I and class II panel reactive antibody (PRA) greater than 85%, which was the reason why she received a desensitisation protocol with plasmapheresis, rituximab and immunoglobulin. She was on the waiting list for more than 5 years without receiving an organ. In February 2014, she had a haemolytic and hypertensive crisis with documented microangiopathic haemolytic anaemia with normal ADAMTS13. A genetic study was completed, revealing a documented mutation in the factor H gene which regulates the Arg257 Cyst complement (a mutation not previously reported and which presented both in this patient and in one of her brothers, both with a history of TMA). For this reason, management was started with eculizumab 900mg; weekly for 4 weeks and then 1200mg fortnightly indefinitely, with prior meningococcal and pneumococcal vaccination. This therapy succeeded in controlling her disease. In November 2014, she joined the kidney transplant protocol at our institution while undergoing treatment with eculizumab. By this time, the patient had a corrected single antigen class I PRA of 87% and class II PRA of 50%. The kidney was transplanted on 21 November 2015. Optimal deceased donor, cold ischaemia time 19h+30min, warm ischaemia time 25min. Compatibility with her donor 2 DR, 1A. She received an immunosuppressant protocol with thymoglobulin, methylprednisolone, belatacept, mycophenolate and re-induction approach with eculizumab. Universal prophylaxis was started with valganciclovir, trimethoprim-sulfamethoxazole, and nystatin (Table 1). The surgery was performed without complications, with immediate function of the kidney graft. Creatinine at day 6 post transplant was 0.8mg/dl, which has been maintained at 18 months of follow-up (Fig. 1).
Treatment protocol used in the kidney transplant.
| Immunosuppression | |
|---|---|
| Induction | Maintenance |
| Thymoglobulin 75mg IV per day for three days | Mycophenolate sodium 720mg po every 12h from day 0 of the transplant |
| Methylprednisolone 500mg IV per day for three days | Prednisolone 50mg po starting from the fourth day after transplant with progressive decrease to 10mg per day |
| Adjustment of the eculizumab dose at the time of the transplant to 900mg IV weekly for four weeks, 1200mg at week 5, and later 1200mg fortnightly | Trimethoprim-sulfamethoxazole 960mg every other day |
| Belatacept: 500mg day 0, 4, 14, 28, 56, 84, and starting in the fourth month 250mg monthly | Valganciclovir 900mg per day for 100 days |
IV: intravenous; PO: orally.
Patients with aHUS who require kidney transplant have a risk of recurrence of the disease that is two times greater than those who received transplants for other reasons, with an incidence as high as 40–60% according to different series, and with a five times higher risk of loss of the kidney graft.5 Among the genetic causes of aHUS, factor H, I, B and C3 mutations are present in 60–70% of cases and patients with the presence of these mutations have a risk of up to 80% of loss of the kidney graft. However, for patients with aHUS secondary to mutations in the membrane cofactor protein (MCP), recurrence of disease after kidney transplant is less common given that the transplanted kidney produces this complement regulator. From there, we glean the importance of a genetic test prior to kidney transplant, which allows for stratification of the risk of recurrence and loss of graft versus the benefit thereof5,6; unfortunately, a percentage of patients do not have an identified genetic mutation and the risk of recurrence is unknown, with there being an association with other less well-documented conditions such as cold ischaemia time, use of calcineurin inhibitors, mTOR, humoral rejection, and infections that trigger or aggravate the process.4,6–8
Currently, eculizumab is one of the recommended therapies to prevent recurrence of aHUS after kidney transplant with satisfactory results in the majority of patients.5,9,10 Zuber et al.10 describe a retrospective multicentre study, 22 kidney transplant patients with aHUS who were administered eculizumab; 9 of them received it as a prophylactic measure before the transplant and one additional dose 24h later, with recurrence occurring in just one patient 14 months after the transplant. The 13 remaining patients received anti-C5 antibody as treatment for recurrence of aHUS post transplant, presenting adequate disease control with greater benefits with the early initiation of the drug and without infectious complications due to encapsulated bacteria. These findings support the use of eculizumab as an aHUS relapse prevention measure and as treatment of recurrences, preventing loss of the graft.8,10–12 It also has a positive effect to prevent and treat humoral rejection in sensitised patients, as was the case of our patient.4,12,13
Also, we must keep in mind that some drugs used as immunosuppressant therapy in kidney transplants are TMA triggers, such as calcineurin inhibitors6,14 and rapamycin inhibitors.15 Therefore, currently other alternatives are being sought to decrease this risk, such as belatacept, which is an immunosuppressant used as maintenance therapy in kidney transplant. This drug is a fusion protein comprising the human IgG1 Fc fragment and the extracellular domain of CTLA-4 (cytotoxic T-Lymphocyte-associated antigen 4), bound with high affinity to CD80 and selectively inhibiting T-cell activation by blocking costimulation14–16 and inducing T cell anergy and apoptosis.2 Belatacept is characterised by the fact it is not nephrotoxic and does not induce TMA,13,17,18 positioning it as one of the drugs with a good safety profile to be used in kidney transplants.19
In the worldwide literature, few cases of the use of belatacept in patients with aHUS post kidney transplant have been documented.15 Merola et al. reported a patient with TMA secondary to the use of calcineurin inhibitors 18h after the transplant, being managed with eculizumab and replacing tacrolimus with belatacept, achieving full recovery of kidney graft function and free of recurrences at two years of follow-up.14 Dedhia et al. published a case of recurrent aHUS due to homozygous factor H mutation 14 days after kidney transplant, which was treated with eculizumab and replacing tacrolimus with belatacept, with recovery of kidney graft function and the patient remaining stable at 2.5 years of follow up.2 To our knowledge, this is the first case reported where belatacept and eculizumab are used in combination as preventative therapy for aHUS recurrence in a kidney transplant patient with a history of factor H mutation.
In conclusion, although kidney transplant in patients with aHUS is frequently associated with relapse and loss of kidney graft, there is currently the possibility of decreasing these risks with the new available therapies such as use of eculizumab and non TMA-inducers such as belatacept, increasing the possibilities of a successful kidney transplant. In the case reported, it was decided to start thymoglobulin induction as it was a highly sensitised patient and eculizumab to prevent the recurrence of aHUS and humoral rejection. As maintenance treatment, it was decided to use belatacept, mycophenolate, and steroids, avoiding potential TMA-inducing drugs such as calcineurin inhibitors, with adequate evolution being achieved to date.
Conflicts of interestThe author John Fredy Nieto-Ríos declares that he has given talks about thrombotic microangiopathies sponsored by Alexion Pharma. All other authors declare that they have no conflicts of interest.
We would like to thank Hospital Pablo Tobón Uribe for allowing to conduct of this case report.
Please cite this article as: Nieto-Ríos JF, Zuluaga-Quintero M, Bello-Márquez DC, Aristizabal-Alzate A, Ocampo-Kohn C, Serna-Higuita LM, et al. Trasplante renal exitoso con protocolo de eculizumab, timoglobulina y belatacept en paciente altamente sensibilizada con síndrome hemolítico urémico atípico por mutación del factor H. Nefrologia. 2018;38:433–437.



