





Podocyte infolding glomerulopathy (PIG) is a condition of uncertain origin, frequently associated with autoimmune diseases. Its specific treatment and clinical course are unknown.
It is characterised by thickening of the capillary walls due to the presence of non-argyrophilic intramembranous bubbles similar to those found in membranous glomerulopathy, but without electron-dense deposits of immune complexes in the ultrastructure, where translucent microspheres generated by invagination of the podocyte cytoplasm into the basement membranes are observed.
ObjectivesGenerally reported in young females patients. To date, few cases in Asian patients have been reported. Our case is the first to be reported in a Latin American Caucasian patient.
MethodsA 38-year-old woman with SLE. In 2014 she presented with nephrotic syndrome empirically treated with corticosteroids (CO) and intravenous cyclophosphamide with good response. She had a relapse in April 2015 with normal renal function and no extrarenal lupus activity, so she was referred to our hospital to be biopsied.
ResultsThe biopsy reported focal segmental glomerular sclerosis without deposits of immune complexes in the immunofluorescence. However, methenamine silver staining revealed clear spaces in the capillary walls accompanied by marked podocyte alterations. On electron microscope study, numerous aggregates of microvesicular and cylindrical ultrastructures bound to the membranes were observed, without evidence of dense deposits, and diffuse effacement of pedicel foot processes, confirming the suspected diagnosis.
ConclusionsThis is the first reported case of what can be considered a new pathological glomerular entity in a Latin American Caucasian patient, whose clinical course and therapy are still unknown.
La glomerulopatía por invaginación podocítica (GIP) es una enfermedad de origen incierto, frecuentemente asociada a enfermedades autoinmunes, de la que se desconoce el tratamiento específico y su evolución.
Caracterizada por engrosamiento de paredes capilares por la presencia de burbujas no argirofílicas intramembanosas similares a las encontradas en la glomerulopatía membranosa, pero sin depósitos de inmunocomplejos electrodensos en la ultraestructura, donde se observan microesferas traslúcidas generadas por invaginación del citoplasma podocítico dentro de las membranas basales.
ObjetivosGeneralmente descrito en pacientes jóvenes de sexo femenino. Hasta la fecha, han sido reportados escasos casos en pacientes de origen asiático. Nuestro caso constituiría el primer reporte en paciente latinoamericano de raza blanca.
MétodosMujer de 38 años con LES. En el año 2014 presentó síndrome nefrótico tratado empíricamente con corticoides (CO) y ciclofosfamida intravenosa (CF) con buena respuesta. Presenta recaída en abril del 2015 con función renal normal y sin actividad lúpica extrarrenal, por lo que es derivada a nuestro hospital para ser biopsiada.
ResultadosLa biopsia informó esclerosis glomerular focal y segmentaria sin depósitos de inmunocomplejos en la inmunofluorescencia, pero con técnica de metenamina plata se detectaron en las paredes capilares, espacios claros acompañados de marcadas alteraciones podocíticas. Al microscopio electrónico, se observaron agregados de ultraestructuras microvesiculares y cilíndricas unidas a las membranas sin evidencia de depósitos densos y borramiento difuso de pies pedicelares, confirmando el diagnóstico sospechado.
ConclusionesReportamos el primer caso de lo que puede ser considerada, una nueva entidad patológica glomerular, en una paciente de raza blanca latinoamericana, cuya evolución y terapéutica aún se desconocen.
Podocytic infolding glomerulopathy (PIG) is a disease of uncertain origin that is commonly associated with autoimmune diseases. Its specific treatment and clinical course are unknown. To date few cases have been reported, all in patients of Asian origin.
In general, the condition is reported in young patients, with a higher prevalence in females.
It is characterised by presenting thickening of capillary walls identified by small non-argyrophilic intramembranous bubbles similar to those found in membranous glomerulopathy, but deposits of electron-dense immune complexes in the ultrastructure are not seen; instead, it is observed the presence of translucent microspheres generated by infolding of the podocytic cytoplasm in the basement membranes.
Our case is the first report in a white Latin American patient.
Case presentationA 38-year-old woman who had a history of systemic lupus erythematosus (SLE) for the past 10 years. In 2014, she presented nephrotic syndrome with intact kidney function and inactive urinary sediment. She was treated with oral corticosteroids and six monthly intravenous pulses of cyclophosphamide, and responded well.
In April 2015, her nephrotic syndrome relapsed, with normal kidney function and no extrarenal lupus activity. She was ultimately referred to Hospital Juan A. Fernández [Juan A. Fernández Hospital] for a kidney biopsy and possible treatment.
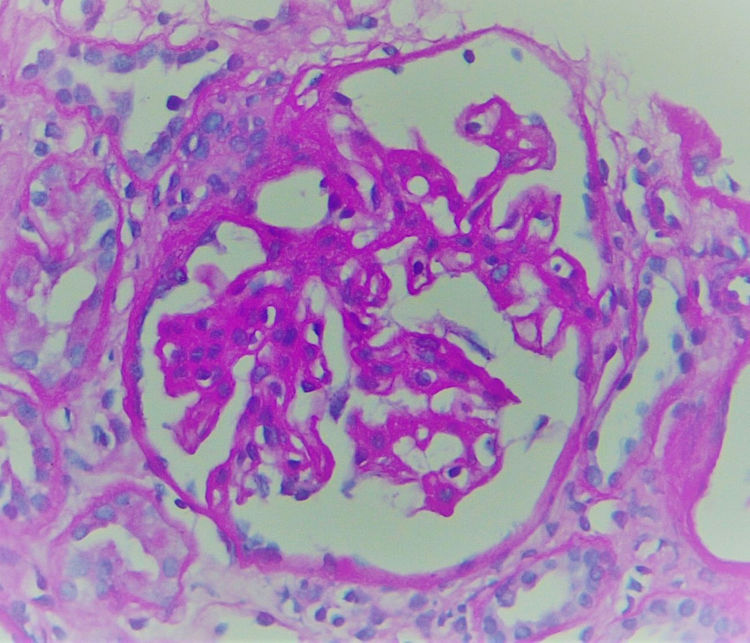
The biopsy included 24 glomeruli, two with global sclerosis and 15 with segmental sclerosis. The results showed expansion of the mesangial matrix and irregular thickening of capillary walls (Fig. 1). Methenamine silver staining reveals non-argyrophilic intramembranous bubbles associated with marked podocytic abnormalities including hypertrophy, vacuolisation, binucleation and detachment (Figs. 2 and 3).
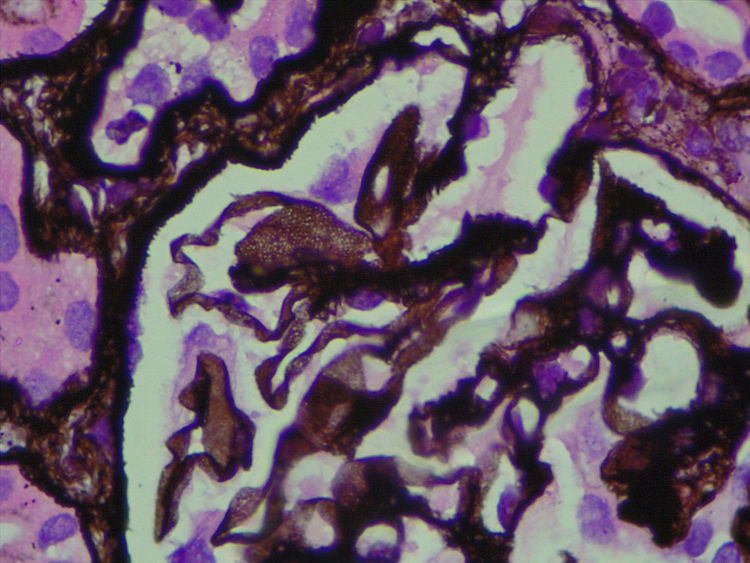
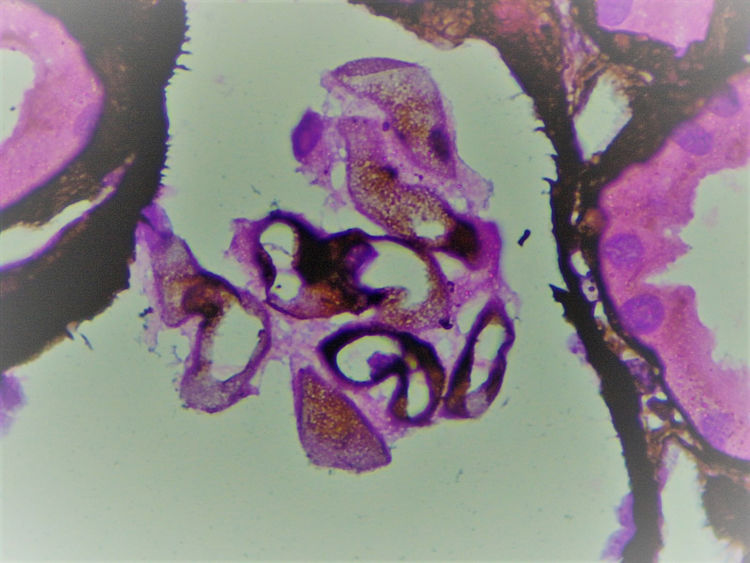
Given that immunofluorescence did not show immune complexes, electron microscopy was essential in order to determine the nature of these clear intraparietal spaces.
Since PIG was suspected, the sample was analyzed by electron microscopy, which showed numerous aggregates of microvesicular and cylindrical ultrastructures attached to the membranes, with no evidence of dense deposits and diffuse podocyte foot process effacement (Figs. 4 and 5). These findings confirmed the suspected diagnosis.
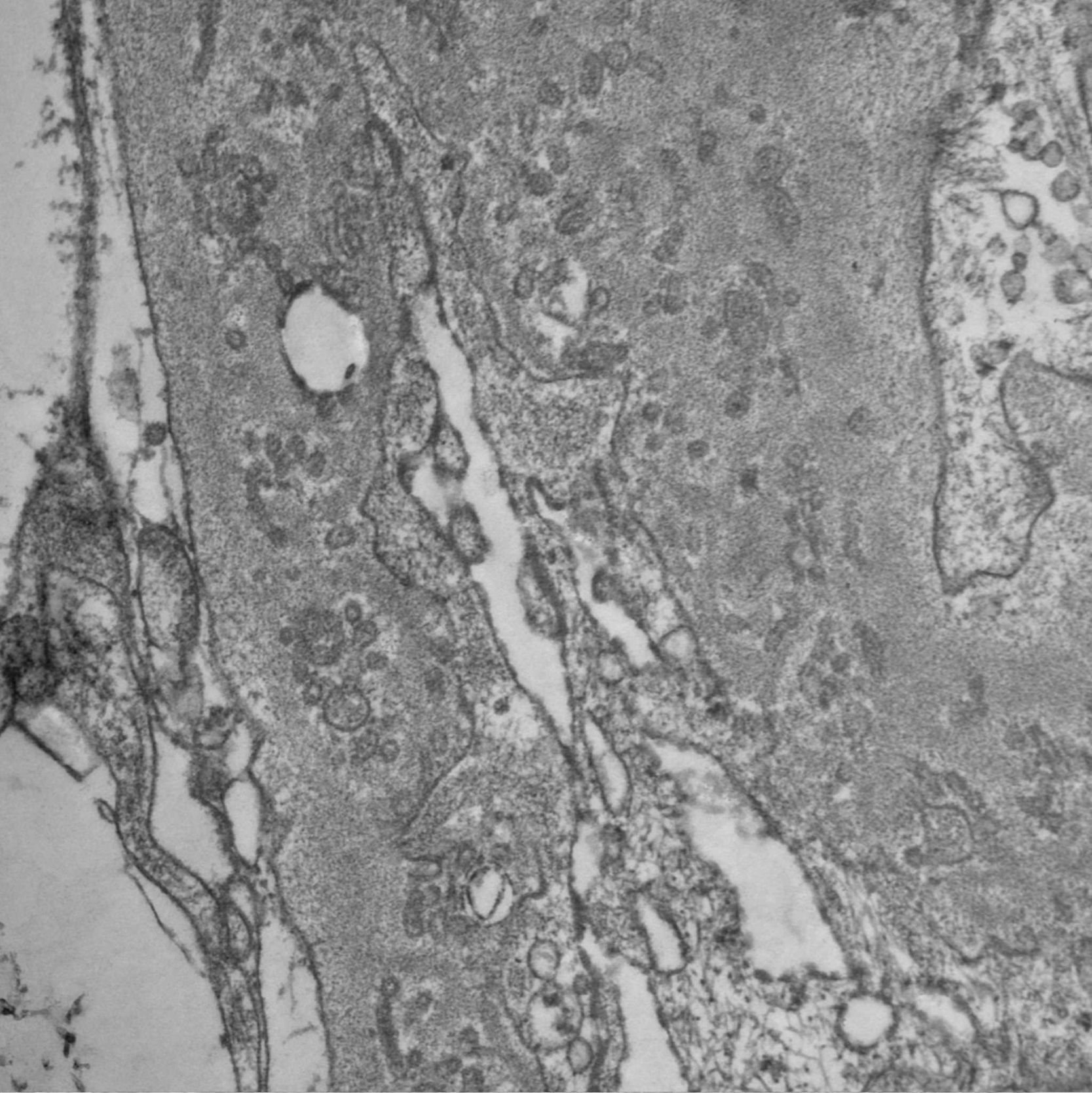
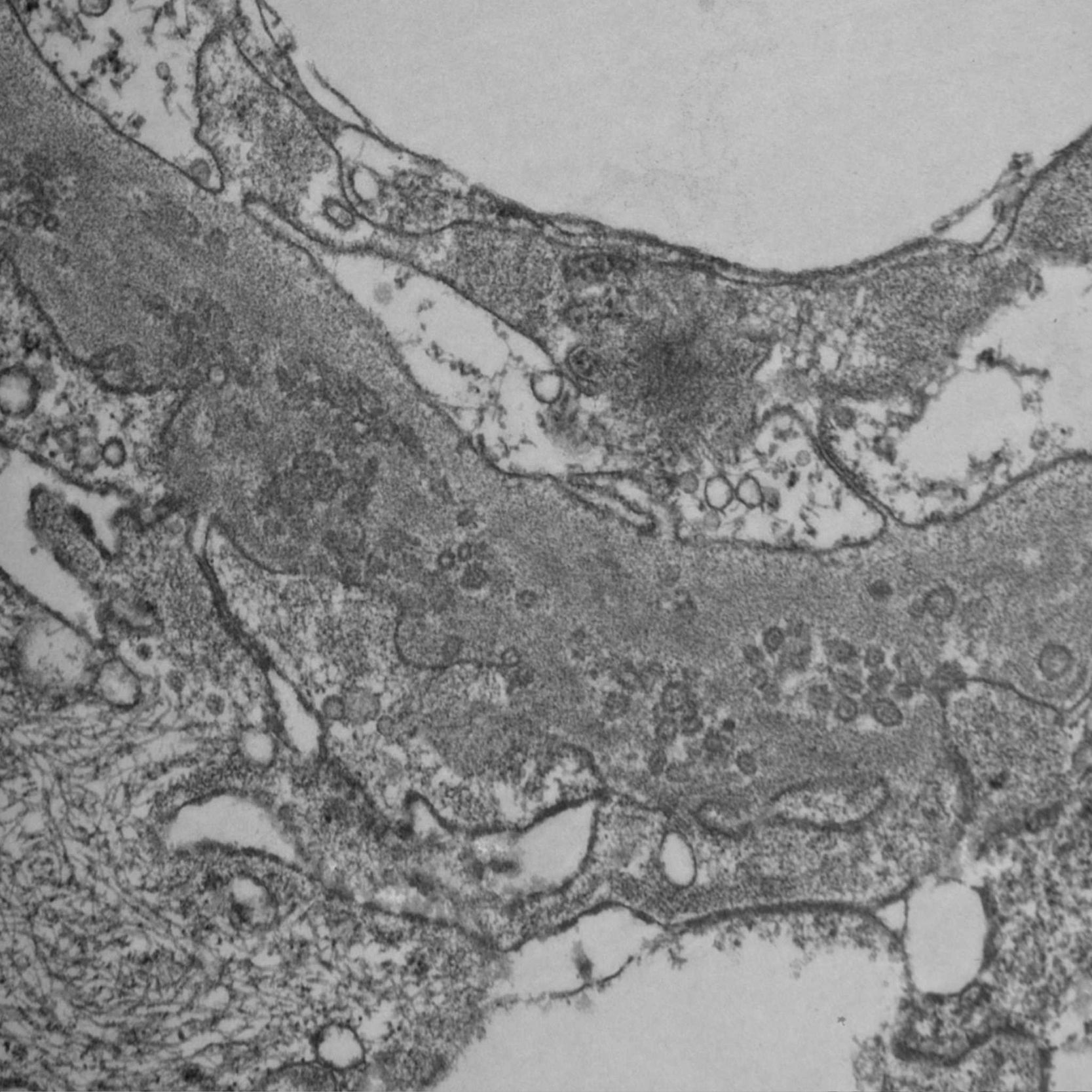
The histopathology findings in this patient are characteristic of an entity initially reported in 1985 as "nuclear pore complex" of the glomerular basement membrane (GBM)1,2 and recently renamed PIG.
The largest series, published by Joh et al.3–6 with 25 cases in Japan in 2008, showed that 17 of these patients also had collagen diseases such as SLE7,8 or Sjögren's syndrome.9 There are also reports of isolated cases of PIG in patients with multiple myeloma,10 primary biliary cholangitis, tumour lysis syndrome,11 Takayasu's arteritis,12 hepatitis B,13 vesicoureteral reflux14 and even cases associated with anaerobic infections.4
It is also associated with patterns of membranous glomerulopathy15,16 and focal segmental sclerosis.17,18
This could indicate that PIG is a result of a non-specific podocyte injury.19
PIG has been divided into two classes depending on whether there is a predominance of "primary infolding" or "microstructures in the GBM". Another classification divides PIG into three categories, depending on the combination of the two forms described above: type A, with primary podocyte infolding only; type B, with microstructures in the GBM plus primary podocyte infolding; and type C, with microstructures in the GBM only.5
The criteria for defining PIG are: non-argyrophilic spaces in the GBM detected by optical microscopy with methenamine silver staining and microspheres or microtubules measuring 50−150 nm in the GBM detected by electron microscopy.20
On immunofluorescence, very weak or negative staining for IgG may be seen along the capillary walls.
It has been mentioned that infolding of the podocytic cytoplasm into the GBM may be a pattern of GBM/podocyte abnormality and not a true disease entity, since it could return to a normal state following withdrawal of the causative stimulus. However, extensive and severe distribution of injury in PIG raises suspicion of intrinsic defects in repair mechanisms.20 The role of podocytes in GBM synthesis, degradation and maintenance has been well documented; therefore, lesions or abnormal conditions in podocytes or abnormalities in the production and degradation of the GBM could lead to podocyte infolding.21
Autoimmune diseases induce failures in biosynthesis and elimination of GBM components by podocytes. In addition, type IV collagen is degraded by metalloproteinases, whereas inhibition of its degradation is caused by metalloproteinase inhibitors. Thus, it is believed that some autoantibodies would affect expression of metalloproteinases, thereby stimulating podocyte infolding in patients with SLE.22
This disease has also been reported in association with diseases involving deposits of immune complexes predominantly with a membranous pattern of injury; other patients show only isolated mesangial and occasionally subendothelial deposits.
Podocytes in the presence of electron-dense immune complex deposits may react, thus introducing their cytoplasm in the GBM in the deposit area.
Podocyte infolding associated with subepithelial deposits is a common finding in membranous nephropathy, especially in stages 2–3.
Preliminary studies in some patients with this condition have managed to identify a circulating antibody that would react with a specific antigen expressed by the podocyte. Its precise identity has not yet been determined, but it is not the same as the M-type receptor for secretory phospholipase A2 (PLA2R) or thrombospondin type-1 domain-containing 7A (THSD7A).
In some glomerulopathies associated with infections, glomerular injury may be expressed by microsphere particles in the GBM, but in these cases it has a focal and segmental distribution, whereas in PIG microspheres they are global and diffuse.23 Therefore, it is recommended that clinicians do not consider the presence of focal podocytic infolding to be a diagnostic criterion for PIG, and instead focus on diffuse microspheres and microtubules in the basement membrane.24
The membrane attack complex (C5-b9) has also been postulated as a podocyte activator, resulting in the lengthening of its cell processes towards the inside of the abnormal GBM. This could account for the presence of intramembranous microstructures, resulting from podocyte and GBM injury, caused for their part by C5-b9 immunoreactants.14
From a clinical point of view, this disease presents in young patients, with a higher prevalence in females, and is usually associated with proteinuria or nephrotic syndrome and a good response to corticosteroid therapy.
Without an ultrastructure study, this condition is often confused with and misdiagnosed as membranous nephropathy.
While most patients present normal kidney function, our patient presented kidney failure due to poor adherence to immunosuppressive therapy and eventual worsening due to sepsis.
ConclusionWe report the first case of PIG in Latin America and in a white female patient. PIG is a disease of uncertain origin commonly associated with autoimmune diseases. As there are few published cases, specific treatments do not yet exist and its clinical course is unknown.
Conflicts of interestThe authors have no conflicts of interest to declare.
Helmut G. Rennke, M.D. Renal Pathology Service, Brigham and Women's Hospital, Pathology Department.
Please cite this article as: Malvar A, Davila P, Ferrari M, Delgado P, Iscoff P, Lococo B, Alberton V, et al. Glomerulopatía por invaginación podocítica; reporte del primer caso en Latinoamérica y revisión de la literatura. Nefrologia. 2020;40:469–473.



