



La enfermedad renal crónica (ERC) se asocia a una elevada morbimortalidad, principalmente de origen cardiovascular. Ello no se puede atribuir únicamente a los factores de riesgo tradicionales, por lo que se han investigado nuevos factores no clásicos. El fósforo (P) se ha postulado como un elemento emergente, y su estrecha asociación con la mortalidad lo ha situado como un factor muy importante en las Guías de Práctica Clínica. La hipótesis clásica del trade off renueva y amplía su protagonismo con el descubrimiento del factor de crecimiento fibroblástico 23 (FGF-23) y Klotho. Estos permiten analizar el diagnóstico y las consecuencias de una retención precoz de P, antes incluso de que se produzca hiperfosfatemia. Además, el aumento del FGF-23 o la disminución de Klotho en estadios iniciales de la ERC podrían convertirse en marcadores no solo de alteraciones del metabolismo óseo-mineral, sino también de la propia ERC y una parte de sus consecuencias sistémicas. De este modo, estudios recientes han descrito la presencia de Klotho como cofactor de la vía de señalización del FGF-23 incluso en el vaso, así como un efecto directo del FGF-23 sobre el cardiomiocito y la hipertrofia ventricular izquierda. Parece que los efectos extraóseos del P, directa o indirectamente a través de FGF-23 o Klotho, van más allá de la calcificación vascular pasiva o activa, imbricándose en procesos de inflamación y estrés oxidativo, entre otros, pudiendo jugar también un papel en procesos de envejecimiento. Por todo ello, un adecuado equilibrio entre estos factores podría convertirse en un futuro en diana terapéutica más allá de la propia ERC.
INTRODUCCIÓN
La enfermedad renal crónica (ERC) se asocia a una elevada morbimortalidad, siendo la primera causa de muerte la de origen cardiovascular (CV). Según datos publicados por el USRDS en su Annual Data Report, la esperanza de vida en EE. UU., por ejemplo, de una mujer de raza blanca de entre 45-49 años de edad es de 35,6 años, mientras que si estuviera en programa de diálisis esta descendería a 7,5 años1. Esta disminución es similar para todos los grupos de edad, sexo, raza, etc. La expectativa de vida del paciente en diálisis es una de las más bajas en medicina, siendo inferior incluso a la de algunas neoplasias comunes. Sin embargo, este riesgo empieza ya antes de la diálisis. Go et al. describen en su trabajo2 que la mortalidad, el número de eventos CV y las hospitalizaciones (con el consecuente aumento del gasto sanitario) crece de modo exponencial con cada estadio de ERC a partir del estadio 3A2. El diagnóstico precoz de la ERC es una de las piezas angulares en los programas de prevención primaria de nuestros pacientes, ya que, según algunos autores, estos pacientes tienen hasta 5-10 veces más probabilidades de morir que de llegar a recibir un tratamiento sustitutivo renal3. Una de las desventajas a la hora de realizar este diagnóstico precoz es que frecuentemente la ERC es asintomática; por ello es de vital importancia el desarrollo de nuevos marcadores de función renal.
El riesgo vital desproporcionado de los enfermos renales no se puede atribuir solo a los factores de riesgo tradicionales, sino posiblemente también a la existencia de múltiples factores de riesgo no clásicos, inherentes a la propia enfermedad, como por ejemplo la anemia, albuminuria, inflamación, estrés oxidativo, malnutrición, entre otros. Las alteraciones del metabolismo óseo-mineral hoy en día son consideradas un componente importante de estos factores de riesgo no tradicionales e imbrica con otros. De este modo, en estudios observacionales los niveles séricos de calcio (Ca), fósforo (P), fosfatasa alcalina (FAL), calcidiol, factor de crecimiento fibroblástico 23 (FGF-23) y/o hormona paratiroidea (PTH) han sido asociados no solo con alteraciones óseas, sino también con calcificación CV, disfunción arterial y, lo que es más importante, aumento de la morbimortalidad (según Block et al. con un riesgo atribuible del 17,5 % en los pacientes en diálisis)4, a pesar de que no existe una prueba prospectiva definitiva de ello. La hiperfosfatemia y especialmente el aumento de FGF-23 son los parámetros que se han asociado de forma más importante, es decir, con mayores riesgos relativos (RR), a mortalidad4, por encima incluso de la PTH, Ca plasmático o FAL4,5.
El exceso de P no solo induce la forma más severa de hiperparatiroidismo secundario (HPS), ya que es un estímulo directo y bloquea todos los mecanismos contrarreguladores, sino que también está relacionado con otros efectos extraóseos que en último término pueden asociarse al aumento de mortalidad («el asesino silencioso»)6. Entre estos efectos, se encuentran los efectos CV directos e indirectos, su asociación con la progresión de la enfermedad renal y el papel central que juega en la calcificación coronaria, valvular y miocárdica.
La hiperfosfatemia siempre se ha considerado de importancia relevante y, aparte de la osteodistrofia renal o su impacto en el sistema CV, tanto P, FGF-23 como Klotho parece que pueden jugar un papel en los fenómenos asociados al envejecimiento y podrían convertirse no solo en marcadores, sino también en dianas terapéuticas para mejorar la supervivencia, incluso más allá de la ERC.
FISIOPATOLOGÍA DEL HIPERPARATIROIDISMO SECUNDARIO Y DETECCIÓN PRECOZ DE LA RETENCIÓN DE FÓSFORO
El P es un mineral indispensable para la vida terrestre: participa en el proceso de fosforilación de proteínas y forma parte del ADN, así como de mensajeros secundarios como el AMP o GMP cíclicos. Ante la escasez de fosfato en la naturaleza, el organismo ha desarrollado sistemas de conservación de dicho mineral, de un modo similar a lo que ocurre con el sodio para la supervivencia fuera del ámbito acuático. Parece más complicado resolver el problema de exceso de fosfato, característico en la ERC, pero, quizás por su alta toxicidad, se han desarrollado también múltiples y elaborados sistemas de eliminación, algunos redundantes, como son la existencia de diferentes fosfatoninas.
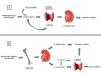
La homeostasis del P se realiza principalmente en el riñón a través de unos transportadores situados en el túbulo proximal (NPT2a, NTP2c) que regulan la excreción/reabsorción de P bajo el mando de numerosos factores metabólicos (principalmente el propio P) y diversas hormonas/fosfatoninas, entre las que destacan FGF-23, su correceptor Klotho y PTH. La vitamina D, el péptido natriurético atrial y los agonistas del óxido nítrico regulan la expresión de NPT2a, y otras fosfatoninas, como FRP-4, MEPE o FGF-7, también regulan la homeostasis del P. La PTH y el FGF-23 reducen la expresión de los receptores NPT2a y NPT2c, disminuyendo la reabsorción renal del P y por tanto aumentando su excreción urinaria. La acción final del FGF-23 se produciría a nivel del túbulo proximal, a través del complejo «receptor del FGF» (FGFR)/Klotho, curiosamente situado en el túbulo distal. Esto implica la probable existencia de un sistema de señalización paracrina que comunique la información desde el túbulo distal al túbulo proximal. El FGF-23 puede también actuar a través de vías independientes de Klotho; por ejemplo, se ha visto cómo actúa directamente en miocardio produciendo efectos deletéreos7. Dado que además de su acción fosfatúrica el FGF-23 aumenta el catabolismo del calcitriol (a través de su acción estimuladora de la actividad de la 24 hidroxilasa renal), parece ser al menos uno de los responsables de coordinar la mineralización del hueso y el balance entre el recambio óseo, las necesidades de fosfato y las de calcitriol. Así, en caso de un aumento de recambio óseo, aumentarían las necesidades de fosfato, disminuiría la producción de FGF-23 por el osteocito, disminuiría la fosfaturia y aumentaría la disponibilidad de calcitriol. En caso de descenso del recambio óseo, se produciría todo lo contrario. Por otra parte, cuando el riñón disfunciona es incapaz de mantener una correcta homeostasis del P y consecuentemente se produce una retención progresiva de P, aumentando también los niveles de FGF-23 de forma precoz, hasta que en estadios más avanzados se incrementan claramente los niveles plasmáticos de P (figura 1). Además, el hueso, que en circunstancias normales actuaría como reservorio de P, es incapaz de tamponar el exceso de mineral en la ERC, empeorando más la retención de P y, consecuentemente, disparando aún más los niveles de FGF-23. El exceso de P circulante puede albergarse entonces en el árbol vascular y los tejidos blandos desencadenando, como también veremos más adelante, procesos de calcificación que empeorarán el pronóstico CV.
Desde la teoría desarrollada por Bricker y Slatopolsky en las décadas de los sesenta-setenta (intact nephron hypothesis), aprendimos que, a medida que se produce la pérdida de nefronas, las restantes se ven obligadas a adaptarse aumentando su fracción de excreción8. Esta idea abrió nuevos campos de investigación. Con relación al P, los trabajos de estos autores dibujaban ya entonces la relación entre el filtrado glomerular (FG) y la reabsorción tubular de fosfato (RTF), de modo que, cuanto más bajo es el FG, más baja es la RTF y más alta la fracción excretada de P (FEF) por nefrona. En la década de los setenta, a raíz de estudios experimentales en animales y humanos, se demostró que la restricción de P en la dieta prevenía el desarrollo de HPS, describiéndose ya entonces el papel central del P en la patogénesis del HPS en la ERC. La retención de P como consecuencia de una disminución del FG produciría una disminución de Ca plasmático que desencadenaría un aumento secundario de la secreción de PTH para restablecer los niveles de Ca y P. Esta es la conocida trade-off hypothesis por la cual el beneficio inicial compensatorio del aumento de PTH, perpetuado y ampliado en el tiempo a cada descenso del FG, provocaría efectos deletéreos sobre el hueso.
Algunos años más tarde, Portale et al. describieron el papel del P indirectamente como inhibidor de la 1-α-hidroxilasa9. La disminución de calcitriol como consecuencia de la retención de P, aunque también por disminución de la masa renal, se postuló como un mecanismo central en el desarrollo del HPS. Ya en los noventa, dos grupos, entre los que destaca el del Dr. M. Rodríguez, describieron el mismo año el efecto directo del P sobre la glándula paratiroidea10,11. Actualmente, la hipótesis del trade-off se rescata y se revitaliza con el descubrimiento de las fosfatoninas y del eje P-FGF-23-Klotho-calcitriol, y no solo han dado paso a nuevas hipótesis temporales durante la progresión de la ERC, sino que también enlazan con el aumento de riesgo CV y vital asociado a la ERC. De este modo, durante las fases iniciales de la ERC, una sobrecarga de P estimularía la síntesis de FGF-23 a nivel de los osteocitos y actuaría en las nefronas restantes aumentando la FEF y manteniendo la normofosfatemia. Hoy en día se cree que el aumento de FGF-23 es reflejo de un aumento crónico de P, de una manera similar a lo que representaría la hemoglobina glicosilada a la glucosa plasmática (figura 2). Por otra parte, las toxinas urémicas parecen inducir la expresión de DNA methyltransferasa (DNMT), una proteína implicada en la silenciación de Klotho a través de hipermetilación12. Debido a estos mecanismos, entre otros, y a la pérdida de masa renal, disminuiría la disponibilidad de Klotho desproveyendo a FGF-23 de correceptor para actuar. Así, con la progresión de la enfermedad renal, FGF-23 perdería su capacidad fosfatúrica, aumentando visiblemente los niveles plasmáticos de P.
Isakova et al. realizaron un estudio con una cohorte de 3879 pacientes en diferentes estadios de ERC y observaron que, ya en estadios muy precoces de la enfermedad (FG 57,8 ml/min/1,73 m2, intervalo de confianza al 95 % 55,4-60,8), el FGF-23 aumentaba, antes incluso de que lo hicieran la PTH, el P o que descendieran los niveles de calcitriol. En el estrato de pacientes con FG entre 50-59 ml/min/1,73 m2, más de la mitad presentaban un aumento de niveles de FGF-23, a diferencia del 3 % de hiperfosfatemia o el 22 % de HPS13. Ello sugiere incluso el potencial uso de FGF-23 como marcador precoz de alteraciones del metabolismo óseo y mineral en la enfermedad renal crónica (CKD-MBD), incluso cuando todavía los niveles de P son normales pero ya puede haberse iniciado una retención corporal de P. A la espera de su introducción en el mercado, parámetros como la FEF podrían utilizarse en la práctica clínica para estimar la presencia de una sobrecarga de P, como recomiendan las guías de la S.E.N.14, antes de la presencia de hiperfosfatemia.
Estudios recientes describen la disminución de Klotho en orina en fases muy precoces de la ERC (estadio 1 y 2), incluso antes de que se produzca el aumento de FGF-23, proponiéndolo como un biomarcador precoz no solo de CKD-MBD, sino también de daño renal15. Los niveles plasmáticos de Klotho, medidos por ELISA, han mostrado resultados algo confusos sobre su disminución en la ERC16. Algunos estudios con modelos animales han investigado la relación de Klotho con fracaso renal agudo por diversas etiologías: isquémico, obstructivo, en contexto de sepsis, hipovolemia o nefrotóxicos. Hu et al. describen como a las 3 h del daño renal por isquemia-reperfusión se puede detectar la disminución de los niveles plasmáticos y urinarios de la proteína Klotho, antes incluso del aumento de N-GAL (Neutrophil Gelatinase-Associated Lipocalin) (5 horas)17. De momento, tendremos que esperar futuros estudios que validen los métodos bioquímicos actualmente utilizados o investiguen otros con mayor sensibilidad.
EJE FÓSFORO-FGF-23-KLOTHO Y MORTALIDAD
Grandes estudios retrospectivos han descrito la hiperfosfatemia como predictor independiente de hospitalización por causa CV, fracturas, muerte súbita, mortalidad CV y global en pacientes en diálisis y prediálisis4,18,19. Block et al. analizaron 40.538 pacientes en hemodiálisis y observaron que los pacientes con niveles séricos de P entre 5-6 mg/dl y 6-7 mg/dl tenían un RR respectivamente de 1,07 y 1,25 veces más de morir que los pacientes con niveles de entre 4-5 mg/dl4. Rodriguez-Benot et al. también describieron en España cómo niveles de P > 5 mg/dl se asociaban de manera independiente con un aumento de la mortalidad en pacientes en diálisis20. Además, en el primer estudio observaron cómo los pacientes que se encontraban en los rangos más bajos de P (< 3 mg/dl) también presentaban un aumento importante de la mortalidad4, probablemente relacionado con malnutrición y/o el síndrome MIA (malnutrición-inflamación-ateromatosis). Así, se ha postulado que controlar los niveles de P solo con restricción proteica podría contrabalancear el beneficio de controlar el P21.
En la población general, los niveles de P también se han asociado a riesgo CV22. Incluso niveles de fosfato en el límite alto de la normalidad (3,5-4,5 mg/dl) se han relacionado con un aumento del riesgo CV, no solo en pacientes afectos de ERC23, sino también en pacientes sin ERC con infarto de miocardio (IAM) previo18. En pacientes con sospecha de enfermedad coronaria y función renal preservada, los niveles de P se han asociado a calcificación y enfermedad coronaria24 y, en niveles altos pero dentro del rango de normalidad, el P se ha asociado también a progresión de la calcificación coronaria25.
Fisiopatológicamente, FGF-23 en un principio se podría considerar que tiene un papel protector sobre la supervivencia al ayudar a mantener la normofosfatemia y prevenir la calcificación vascular. Recientemente se ha descrito la presencia de Klotho en células vasculares26 como posible cofactor en la vía de señalización de FGF-2327. El mismo grupo ha demostrado cómo la deficiencia de Klotho en ERC puede ser restaurada, desenmascarando efectos anticalcificantes del FGF-2327. Sin embargo, con la progresión del daño renal, el exceso de FGF-23 biológicamente activo producido en respuesta a la hiperfosfatemia podría tener potenciales efectos adversos directos o indirectos. De este modo, el FGF-23 contribuye a la disminución de calcitriol y otros múltiples efectos off target descritos en estudios observacionales, como: disfunción vascular, ateroesclerosis o hipertrofia ventricular izquierda (HVI), potencialmente responsables del aumento de mortalidad. Marchais et al. observaron cómo pacientes en diálisis hiperfosfatémicos presentaban mayor presión diastólica y media, mayor índice cardíaco y mayor relación pared/luz carotídea que los pacientes con niveles normales de P28. El conjunto de anormalidades hemodinámicas podría resultar en un aumento de la masa ventricular izquierda y comprometer así el flujo coronario durante la diástole como cualquier otra situación que favorezca la rigidez vascular.
Sin embargo, existe actualmente un debate abierto sobre si el FGF-23 es nocivo per se o es tan solo un mecanismo adaptativo en respuesta a la hiperfosfatemia, pudiendo ser esta última la causante del aumento de mortalidad y toxicidad CV29. Como se ha mencionado antes, Gutiérrez et al. describieron cómo el FGF-23 se asocia de manera significativa a un aumento de mortalidad en pacientes en diálisis incluso tras ajustar para parámetros como P sérico, PTH o vitamina D30. Esta asociación ocurre también después de un ajuste multivariante en estadios previos a la diálisis (ERC 2-4), donde se ha descrito que la relación es más intensa incluso que con los factores de riesgo tradicionales, la proteinuria o el FG estimado. En otro gran estudio, realizado por Kendrick et al., se analizó una muestra de 1099 pacientes con FG medio < 18 ml/min/1,73 m2 y se confirmó una asociación independiente entre eventos CV (objetivo principal compuesto de IAM, amputación e ictus) y niveles de FGF-2331. Actualmente la relación del FGF-23 con la HVI es una de la hipótesis más plausibles para justificar su asociación con la mortalidad independiente de P. FGF-23 parece inducir HVI en la ERC a través de un mecanismo directo independiente de Klotho7. Estudios experimentales con ratones han demostrado cómo FGF-23 hipertrofia de manera patológica el cardiomiocito a través de la activación del receptor para FGF-23 mediante la vía de señalización calcineurina-NFAT (Nuclear Factor of Activated T Cells) y de manera independiente de Klotho. En ratones deficientes para Klotho, la inyección intraventricular o intravenosa de FGF-23 favorece la HVI y la administración de bloqueadores de FGFR la redujo significativamente sin cambios en los valores de presión arterial7. Sin embargo, la neutralización con anticuerpos de FGF-23 mejoró el HPS en un modelo animal pero empeoró la supervivencia, probablemente en relación con el aumento de P y calcificación vascular29. Así pues, es posible que el efecto adaptativo de FGF-23 ante la retención de P pueda ser beneficioso al disminuir la sobrecarga de P, pero que pueda resultar deletéreo en presencia de niveles excesivamente elevados o sobre aquellos órganos en los que no se expresa su correceptor Klotho.
Klotho es una de las diosas más antiguas de la mitología griega; junto a sus dos hermanas, trabajaba el hilo de la vida, y estaba en sus manos el destino del hombre y de los dioses. El mismo nombre se dio a un gen que codificaba para una fosfatonina y se observó que cuando los ratones eran deficientes para dicho gen desarrollaban osteopenia, calcificaciones vasculares y una serie de desórdenes relacionados con el envejecimiento acelerado que acortaba su esperanza de vida: esterilidad, enfisema, hipoglucemia, sordera, ataxia, fragilidad cutánea, etc.32. Los ratones FGF23 -/- presentan un fenotipo similar a los Klotho -/-32,33. La relación de Klotho con la supervivencia podría explicarse: 1) a través de la inhibición de señales inflamatorias que producirían una resistencia al estrés oxidativo, como veremos más adelante; 2) por las alteraciones del metabolismo mineral que representa su deficiencia: aumento del P y calcitriol, hipercalciuria e hipofosfaturia. Esta última hipótesis viene apoyada por estudios experimentales con ratones donde se ha observado que, al restringir el P de la dieta, mejora la expresión de Klotho en sus riñones, y presentan menores calcificaciones ectópicas (costales y en riñón)34. De un modo similar, en un estudio para establecer detalladamente el fenotipo del síndrome de progeria de Hutchinson-Gilford (envejecimiento prematuro), se describió que 8 pacientes de los 15 estudiados presentaban niveles elevados de P sérico (5,5 ± 0,2 mg/dl), con una función renal normal y una FEF inferior al 20 %35.
EJE FÓSFORO-FGF-23-KLOTHO Y CALCIFICACIÓN VASCULAR
Además de los mecanismos señalados con anterioridad, la calcificación parece contribuir de forma importante al riesgo CV. Es altamente prevalente en la ERC, siendo en estadios iniciales el doble que en la población general y hasta 9 veces más prevalente en estadios avanzados. Todo ello hace actualmente del modelo animal urémico un paradigma de estudio de calcificación vascular extensible a otras especialidades del ámbito CV.
Clásicamente, se ha considerado que existen dos tipos de calcificaciones en función del lugar de depósito mineral, la calcificación intimal y la medial, ambas presentes en la ERC. La calcificación intimal se relaciona con procesos de aterosclerosis (placa de ateroma) y está localizada principalmente en arterias coronarias, aumentando el riesgo de isquemia, erosión y ruptura de la placa. La calcificación medial, relacionada con procesos de arterioesclerosis, se localiza principalmente en arterias de mayor compliancia (arterias elásticas) y es causante de la rigidez arterial y el aumento de presión de pulso comúnmente observado en los pacientes con ERC. Estos cambios hemodinámicos tienen como consecuencia la HVI, compromiso del llenado coronario e insuficiencia cardíaca a largo plazo.
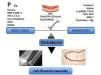
En la ERC se produce una transformación activa de células de la musculatura lisa vascular (CMLV) a células similares a osteoblastos. Los procesos de calcificación vascular se producen también por un desequilibrio entre promotores (Ca, P, BMP [proteína morfogénica ósea] 2, BMP-4, RANKL [ligando de receptor activador para el factor nuclear κ B], interleucina 6 [IL-6], factor de necrosis tumoral α [TNF-α]), activación de factores de transcripción como CbFα1/RunX2/Osterix, mediadores de la inflamación, entre los que se encuentra NFκB (factor nuclear κB), e inhibidores de dicho proceso. Algunos de los inhibidores serían la fetuina A en plasma, MGP (proteína gla de matriz), osteopontina, osteoprotegerina, BMP-7 o pirofosfatos (figura 3). También se han descrito como potenciales factores que favorecen la calcificación de la CMLV en medio urémico el estrés oxidativo, los AGE (productos finales de glicación avanzada), la hipertensión arterial, la enfermedad ósea adinámica o la dislipemia. La enfermedad ósea, como se ha comentado anteriormente, adinámica no permitiría tamponar los excesos de Ca y P por parte del hueso. También el uso de algunos fármacos habituales en la práctica clínica de los pacientes en diálisis podría favorecer la calcificación (quelantes del P cálcicos, vitamina D a dosis altas o warfarina) y otros podrían hipotéticamente retrasar su progresión (quelantes del P no cálcicos, calcimiméticos, bifosfonatos, inhibidores del sistema renina-angiotensina, estatinas, antagonistas del Ca o la vitamina K). En modelos experimentales se observa que los activadores selectivos del receptor de vitamina D, como el paricalcitol, podrían tener menos efectos procalcificantes que el calcitriol. Este efecto podría ser atribuido no solo a que el paricalcitol se asocia con niveles menores de Ca x P, sino también al hecho de que paricalcitol disminuye la expresión de factores osteogénicos como RunX2/Cbfα1, Osterix, Msx2, disminuye la expresión de BMP-2 y los niveles nucleares de β-catenina con sus genes relacionados, previniendo la transformación de las CMLV en osteoblastos36,37.
Como hemos visto, el P facilita la calcificación de los vasos también favoreciendo la formación de núcleos de cristales de bioapatita. El paso de P al interior de la célula se realiza a través de cotransportadores Na-P (NTP). El NTP-III (Pit-1 y Pit-2) se expresa en las células del musculo liso (Pit-1 > Pit-2). El P atraviesa la membrana, a través de Pit-1, y una vez dentro induce formación de vesículas mineralizantes y la transformación fenotípica osteocondrogénica. No debemos olvidar la importancia del Ca y que algunos autores como Yang et al. demostraron que el Ca sobrerregula también la expresión de Pit-1, produciendo un efecto sinérgico procalcificante junto a la hiperfosfatemia38. Del mismo modo, en estudios experimentales in vivo se ha observado cómo la acidosis puede disminuir la calcificación de tejidos blandos39.
También en la calcifilaxis (arteriolopatía urémica calcificante), patología que registra altas tasas de mortalidad, la hiperfosfatemia estaría implicada al inducir la calcificación de las arteriolas, multiplicando por 3,5 el riesgo por cada aumento de 1 mg/dl de P sérico40.
EJE FÓSFORO-FGF-23-KLOTHO, INFLAMACIÓN Y ESTRÉS OXIDATIVO
En las últimas décadas, el proceso aterosclerótico ha pasado de ser puramente metabólico a resultar mucho más complejo, incluyendo fenómenos inflamatorios. La inflamación es una condición frecuente en la ERC, sobre todo en estadios terminales de la enfermedad (> 50 % de pacientes) que podría contribuir en parte a justificar el alto riesgo CV que presentan estos pacientes. Diversos factores, como la carga genética, el estrés oxidativo, la disminución del aclaramiento de citocinas proinflamatorias (como las mencionadas TNF-α, IL-6) o disfunción autonómica, podrían contribuir al estado inflamatorio en la ERC.
El P está implicado en la regulación de muchas vías inflamatorias a través de procesos de fosforilación, por lo que algunos autores han sugerido que la hiperfosfatemia podría activar directamente la cascada inflamatoria41. En este sentido, Navarro et al. publicaron un elegante estudio en pacientes con ERC estadio 3-4 sin historia de enfermedad CV ni tratamiento con suplementos cálcicos, quelantes del P o vitamina D, y observaron cómo los niveles de PCR (C-Reactive Protein) ultrasensible e IL-6 eran superiores en los pacientes con P ≥ 5 mg/dl (p < 0,001). El P se correlacionó de una manera significativa e independiente con la PCR (r = 0,35, p < 0,01) e IL-6 (r = 0,38, p < 0,0001)42.
El P es uno de los sustratos de la fosforilación oxidativa y juega un importante papel en la regulación del potencial de membrana mitocondrial y la producción de radicales superóxido. El P después de entrar a la célula a través de Pit-1 estimularía la producción de radicales superóxidos que activarían la vía del NFκB que, una vez translocado al núcleo (a través de p65), se uniría a secuencias específicas de ADN para transcribir programas osteogénicos43. El estrés oxidativo podría aumentar la transcripción de RunX2 y MsX2, entre otros factores de mineralización vascular43,44.
Con relación a la inflamación, también se han descrito algunas acciones enzimáticas de Klotho alternativas a su versión transmembrana como cofactor de la señalización de FGF-23. De este modo, la forma circulante de Klotho (perteneciente a la familia de las β-glicosidasas) actúa como un factor humoral inhibiendo cascadas de señalización intracelulares, como la de la insulina/factor de crecimiento insulínico tipo 1, y otras vías que tienen como consecuencia la formación de radicales que promueven el envejecimiento celular45. Así, Klotho, a través de aumentar la resistencia al estrés oxidativo, podría aumentar la longevidad de algunas especies.
EJE FÓSFORO-FGF-23-KLOTHO Y PROGRESIÓN DE LA ENFERMEDAD RENAL
El P podría promover la progresión de la ERC a través de mecanismos todavía no bien conocidos. Algunos autores postulan la hipótesis de la precipitación-calcificación, basada en el depósito de cristales de fosfato cálcico en la célula tubular e intersticial, provocando daño celular y proliferación de fibroblastos. También se sabe que la sobrecarga de P daña el podocito en animales de experimentación, pudiendo explicar la relación descrita entre el P y la gravedad de la proteinuria. En este sentido, estudios recientes describen cómo una dieta rica en P se asocia con un aumento de la expresión renal de la enzima de conversión de la angiotensina en ratas nefrectomizadas, aumentando el daño tisular.
En humanos, se analizaron de forma retrospectiva en el estudio REIN 331 pacientes con proteinuria y ERC, observándose que el efecto antiproteinúrico del ramipril se atenuaba significativamente a medida que aumentaban los niveles de P46. En la población general, se ha descrito incluso que los niveles de P en el cuartil alto de la normalidad se asocia de manera independiente a la presencia de microalbuminuria.
Asimismo, Caravaca et al. observaron también cómo existía una correlación lineal entre la variación del FG y el P sérico en pacientes con ERC avanzada47. En este mismo trabajo se observó que los pacientes tratados con diuréticos mostraban una concentración media de P sérico significativamente más elevado47. Otros autores describen que, por cada mg/dl de aumento del P, el FG disminuye significativamente 0,154 ml/min/mes48. En el estudio anterior se observó cómo la velocidad de deterioro de función renal era más intensa cuanto más alta era la función renal residual basal, sugiriendo que el P podría jugar un papel independiente en la progresión de la ERC47.
El FGF-23 podría ser también un nuevo marcador de progresión renal, como indican algunos estudios donde se ha visto que existe una correlación inversa entre sus niveles y el descenso de FG. Otros autores sugieren que la combinación de elevación de FGF-23 con disminución de los niveles de calcidiol proporciona la mejor estratificación del riesgo de progresión de la función renal. Sin embargo, en pacientes con ERC avanzada (FG < 30 ml/min/1,73 m2), la relación entre FGF-23 y ERC terminal se pierde al ajustar por FG, ya que este último es el principal determinante en la progresión de la ERC49. Se necesitarán futuros estudios para dilucidar si FGF-23 actúa como biomarcador de ERC o posee un mecanismo directo en la progresión de la enfermedad. Como se ha mencionado antes, también se ha descrito que Klotho podría tener un papel renoprotector, como sugieren algunos estudios experimentales, reduciendo la apoptosis en fracasos renales agudos de origen isquémico.
CONCLUSIONES
En esta revisión hemos visto cómo en los últimos años se han descrito múltiples efectos del P más allá de la fisiopatología de la osteodistrofia renal y del HPS. Su papel emergente como factor aterogénico, procalcificante y su asociación con aumento de mortalidad han hecho que algunas sociedades nefrológicas, entre ellas la Sociedad Española de Nefrología14, se hayan posicionado en sus guías de práctica clínica hacia la consecución de unos niveles séricos de P estrictamente normales en los pacientes afectos de ERC sea cual sea su estadio, incluyendo los pacientes en diálisis, y siempre que las medidas para conseguirlo sean razonables, modificando recomendaciones como «hacia la normalidad» de las academicistas KDIGO referidas a los pacientes en diálisis50. Sin embargo, la hiperfosfatemia es un marcador tardío de retención de P. La disminución de Klotho y el aumento de FGF-23 parecen ser eventos precoces responsables del desarrollo de HPS y de efectos sistémicos más allá del hueso y de la calcificación vascular. El inicio de terapia cuando ya ha aparecido la hiperfosfatemia o la restricción en ficha técnica de los quelantes no cálcicos que recomiendan su uso solo cuando el P es > 5,5 mg/dl (> 1,78 mmol/l) queda a nuestro entender desfasado a la luz de los conocimientos de la fisiopatología actual, pues es posible que una maniobra beneficiosa y simple para paliar los efectos sistémicos de la sobrecarga de P sea intervenir precozmente. A la espera del desarrollo de nuevas dianas terapéuticas, el uso de la FEF puede resultar útil para dirigir el tratamiento precoz, siendo recomendable iniciar terapia cuando la RTF esté significativamente disminuida (p. ej. < 80 %). Sin embargo, aunque estas directrices parecen razonables, debemos admitir que no existen resultados en estudios prospectivos que confirmen la utilidad de esta práctica. Todos estos avances en la fisiopatología podrían repercutir en el futuro no solo en forma de nuevas estrategias y terapias sobre el riesgo CV y progresión de la ERC, sino también sobre los procesos de envejecimiento acelerado en la ERC y mejoría de la supervivencia, potencialmente aplicables a la población general.
En la ERC, la dieta, la diálisis y los quelantes del P son alternativas terapéuticas complementarias de las que disponemos actualmente para tratar la retención de P y/o la hiperfosfatemia. Este capítulo ha querido ser la introducción general de un suplemento dedicado a un captor del P como carbonato de lantano, en el que se desarrollan aspectos como la seguridad, la adherencia y coste-eficacia del fármaco, así como el control que ejerce sobre la retención de P o la salud ósea y la relevancia que tienen los quelantes del P sin Ca en el seno de la ERC como enfermedad de extremo riesgo CV.
Agradecimientos
Fondos FEDER RETIC REDINREN del FIS RD12/0021.
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.

Figura 1. Cambios bioquímicos en la fisiopatología del hiperparatiroidismo secundario

Figura 2. Esquema del trade off clásico (A) y actualizado (B)

Figura 3. Proceso de calcificación vascular: desequilibrio entre inductores e inhibidores


