




Nephropathic cystinosis is an autosomal recessive lysosomal storage disorder that is characterised by the accumulation of the amino acid cystine in several body tissues due to a mutation in the CTNS gene, which encodes the cystinosin protein. The aim of this study was to sequence the coding exons of the CTNS gene in five different Jordanian families and one family from Sudan with nephropathic cystinosis.
MethodsProbands initially presented with Fanconi syndrome symptoms. An eye examination showed the accumulation of cystine crystals in the cornea by the age of 2 years, suggesting cystinosis. All of the coding exons and flanking intronic sequences and the promoter region of the CTNS gene were amplified using polymerase chain reaction and subjected to sequencing.
ResultsNone of the probands in this study carried the European 57-kb deletion in the CTNS gene. Seven variants in the coding and promoter sequence of the CTNS gene were identified in the probands of this study. Two of these variants were a CTNS mutation that was previously identified in a heterozygous genotype in two different patients of European descendant. The two mutations were c.829dupA in exon 10 and c.890G>A in exon 11. The proband of family 2 was compound-heterozygous for the two mutations.
ConclusionThis study is the first molecular study of infantile nephropathic cystinosis in Jordan. We successfully identified the causative CTNS mutations in Jordanian families. The results provide a basis for the early detection of the disease using molecular tools in a highly consanguineous Jordanian population.
La cistinosis nefropática es una enfermedad de almacenamiento lisosómico autosómica recesiva que se caracteriza por la acumulación del aminoácido cistina en varios tejidos del cuerpo. Ello se debe a una mutación en el gen CTNS, que codifica la proteína cistinosina. El objetivo de este estudio fue secuenciar los exones codificantes del gen CTNS en cinco familias jordanas afectadas por la cistinosis.
MétodosLos casos índice se presentaron inicialmente con síntomas del síndrome de Fanconi. Exámenes oftalmológicos revelaron la acumulación de cristales de cistina en la córnea a la edad de 2 años, lo que sugería la existencia de cistinosis. Todos los exones codificantes, las secuencias intrónicas flanqueantes y la región promotora del gen CTNS se amplificaron mediante reacción en cadena de la polimerasa y fueron objeto de secuenciación.
ResultadosNinguno de los casos índice de este estudio presentaba la deleción 57-kb europea en el gen CTNS. Se identificaron siete variantes en las secuencias codificante y promotora del gen CTNS en los casos índice de este estudio. Dos de estas variantes eran una mutación del gen CTNS previamente identificada en un genotipo heterocigótico de dos pacientes de descendencia europea. Las dos mutaciones eran c.829dupA en el exón 10 y c.890G>A en el exón 11. El caso índice de la familia número 2 era heterocigótico compuesto respecto a las dos mutaciones.
ConclusiónEl presente estudio es el primer estudio molecular sobre cistinosis nefropática infantil en Jordania. Logramos identificar con éxito las mutaciones en el gen CTNS causante en familias jordanas. Los resultados sirven de base para la detección precoz de esta enfermedad mediante herramientas moleculares en una población jordana marcadamente consanguínea.
Cystinosis is an autosomal recessive disease with a 1/100,000- to 1/200,000-live-birth prevalence.1 A combination of physical mapping and linkage analysis successfully linked cystinosis to a region on chromosome 17p13, and the CTNS gene was positionally cloned and sequenced, with several mutations identified in families with cystinosis.2,3 The CTNS gene consists of 12 exons, of which exons 1 and 2 and the first 19 nucleotides of exon 3 constitute the non-coding region of the gene.3
According to the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php – assessed 2014 Jul 11), more than 100 mutations have been characterised thus far in the CTNS gene. Recent studies from the Middle East have identified several CTNS mutations, some of which were new.4–10 The c.681G>A CTNS mutation is the most prevalent mutation in Middle Eastern countries, whereas the founder 57-kb deletion mutation is the most common mutation that is detected in northern European populations.3,4,7,10,11 Both the precise telomeric and centromeric breakpoints for the 57-kb deletion were mapped by sequencing more than 200kb harbouring the CTNS gene using overlapping BAC clones that were constructed from chromosome 17.12 Several molecular assays were developed to screen for the 57-kb deletion mutation, including multiplex PCR analysis using primer pairs within and outside of the deletion breakpoints and FISH analysis.13,14 Worldwide, the mutation spectrum of the CTNS mutation is heterogeneous, as several mutation types have been described in Italian, French, German and Swiss origin, Mexican, Indian and Thai and Spanish populations.15–22
Clinically, cystinosis is classified into three different types depending on the disease severity and the age of manifestation. These forms include infantile (OMIM: 219800) and juvenile (OMIM: 219900) nephropathic cystinosis and the ocular non-nephropathic (OMIM: 219750) form. The infantile form is the most common type of the three clinical variants of cystinosis and presents in the first year of life with the renal tubular electrolyte imbalance termed “Fanconi syndrome”; the patient reaches end-stage renal disease by the age of 10 years if not treated with cysteamine.23
The CTNS gene encodes a seven-transmembrane-domain (TM) lysosomal protein called cystinosin, whose localisation to the lysosome is guided by two sorting signals; a tyrosine-based sorting signal that is located at the c-terminus of the protein and a novel sorting signal that is located between the fifth and sixth transmembrane domains of the protein.24 The deletion of either one of the two sorting signals results in partial mislocalisation of the cystinosin protein to the plasma membrane, and a lysosomal signal is also observed.24 A whole COS-7 cell functional assay demonstrated that cystinosin is a proton-driven cystine transporter from the lysosome; these findings form a foundation for understanding the molecular pathogenesis of cystinosis, on which several genotype-phenotype studies have been published.18,25,26
The promoter of the CTNS gene was functionally characterised using several chloramphenicol acetyltransferase (CAT) gene reporter constructs.27 The basal CTNS promoter was defined as the region between nucleotide −316 to +1.27 Three regulatory cis-elements were functionally characterised in the CTNS promoter, two of which harboured the Sp-1 transcription factor binding site.27 A promoter mutation (−295G>C) in the distal Sp-1 binding site was previously identified in one cystinosis patient.27 In another functional study, CTNS promoter constructs harbouring the −294 T mutant allele of the Sp-1 binding site also showed reduced activity.28 Here, we report for the first time the molecular basis for cystinosis in Jordanian patients.
MethodsProbands and familiesThis study was approved by the joint institutional review board committee of the Jordan University of Science and Technology and King Abdullah University Hospital. Consent was obtained from the patients’ parents and from the parents themselves for family 6. All of the patients were Jordanians except for the proband of family 5 who was Sudanese.
In family 1, the proband was a 4-year-old female born to first-cousin parents. At the age of 2 years, she began to have recurrent urinary tract infections. She was diagnosed with hypothyroidism and also found by ultrasound to have nephrocalcinosis. At the age of 4 years, she showed signs of Genu Varum and bowed legs and was found to have rickets. She was then referred to us, where she was diagnosed with Fanconi syndrome. Slit lamp examination confirmed the presence of crystals in the cornea, suggesting cystinosis. DNA samples from her sister, grandmother (father's side), and parents were available for testing.
In family 2, the proband was 5 years and 3 months old and born by normal vaginal delivery to second-cousins parents with a birth weight of 2.18kg. She was found to have failure to thrive, hypokalaemia, hyponatraemia, hypochloraemia, polyuria and polydipsia, suggesting Fanconi syndrome. An X-ray at 18 months showed signs of rickets, and a slit-lamp examination of the cornea confirmed the diagnosis of cystinosis. She died at the age of 7 years after receiving renal dialysis for 3 months. The family had a history of one sibling death at the age of 1 year and 7 months with similar symptoms. Samples from the brother and parents were available for testing.
The proband from family 3 was born to second-cousins parents and was a result of normal vaginal delivery with a birth weight of 2kg. During the neonatal period, the mother observed the proband having an attack of febrile illness and drink excessive milk. The child's illness was associated with constipation and with failure to gain weight. At the age of 6 months, she was admitted to the hospital to be investigated for failure to thrive and was found to have rickets, hypophosphataemia, hypokalaemia, metabolic acidosis and glucosuria, suggesting Fanconi syndrome. Then, a slit lamp examination showed negative result for corneal cystine crystals. At the age of 3 years, the slit lamp examination was repeated and confirmed the presence of cystine crystals, and the diagnosis of cystinosis was made. Cysteamine treatment was refused by the patients’ parents, and by the age of 3 years and 8 months, the patient began to show symptoms of renal failure. Samples from the brother and parents were available for testing.
In family 4, the proband was admitted to the hospital at the age of 6 months with failure to gain weight, polyuria and polydipsia. The parents were second-cousins with a positive family history of cystinosis. Physical examination revealed that the baby was in the 5th percentile for both height and weight. Lab tests showed evidence of hypokalaemia, hypophosphataemia, metabolic acidosis, glucosuria and proteinuria, along with radiological changes of rickets. The ophthalmological exam was normal at 6 months of age. The leucocyte cystine level was 2.0nmol half-cystine/mg of protein at the age of 1 year and 7 months with typical corneal crystals. Oral cysteamine therapy was initiated shortly thereafter, and patient died at the age of 10 years as a result of end-stage renal failure.
In family 5, the proband was an 11-month-old male infant that was born to first-cousins of Sudanese nationality. There was a positive family history with unexplained deaths of 3 siblings during infancy. The proband started at the age of 4 months to have recurrent episodes of vomiting and diarrhoea, requiring admission to the hospital on several occasions. Upon X-ray, he was found to have widened wrists, suggesting active rickets. Investigations showed persistent hypokalaemia, hypophosphataemia with acidosis and a normal anion gap. Urine testing for amino acids showed generalised aminoaciduria. Based on these findings, he was diagnosed with Fanconi syndrome and was started on appropriate supplements. An ophthalmological evaluation confirmed the presence of crystals in the cornea suggestive of cystinosis. Samples from the parents were available for testing.
In family 6, the parents who were second-cousins sought genetic counselling because three of their children's were died of cystinosis before this study started. The children's died at age of 2 months (son), 3 years (daughter) and 2 years (second daughter). Thus, DNA samples were unavailable from the children's. The clinical record for the second daughter was available at the Paediatric department of King Hussein Medical Centre in Amman. She was admitted to hospital at age of 1 year and 6 months with clinical signs resembling Fanconi syndrome. At age of admission a slit lamp examination showed negative result for corneal cystine crystals but her X-ray showed radiological changes of rickets. Despite the patient was on regular electrolyte replacement and cysteamine treatments, here kidney function deteriorated and died at the age of 2 years.
Mutation analysisThe genomic DNA from patients and their available family members was extracted from whole blood using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA). The polymerase chain reaction was performed in an iCycler Thermal Cycler (Bio-Rad, Hercules, CA, USA). The 57-kb deletion was evaluated using a previously described method.13 Briefly, two independent multiplex PCR reactions were performed: the first multiplex PCR reaction including the primer pairs LDM1 and D17S829 and another reaction including the LDM2 and exon 4 primer pairs. The upstream and downstream PCR primers and reaction conditions to amplify CTNS exons 3-12 and the flanking intronic sequences were essentially as described by Town et al.3 The promoter primers were designed using the Primer3 algorithm (http://frodo.wi.mit.edu/primer3) according to GenBank accession number NM_001128425.1 and had the following sequences: forward, [5′-TGCCAATCTTTCAGCCACAC-3′] and reverse, [5′-TCTTAGACGGACAGAGCGC-3′]. All of the PCR products were subjected to bi-directional sequencing using the Big-Dye Terminator v3.1 Cycle Sequencing Kit from Applied Biosystems before being applied to an ABI PRISM 310 Genetic Analyser (Applied Biosystems, Foster City, CA, USA). The sequence data were compared to the normal CTNS gene sequence (GenBank accession number for the c-DNA: NM_004937.2; GenBank accession number for genomic: NG_012489.1) using the ChromasPro 1.34 (Technelysium Pty Ltd, Australia) software package. Sequence nomenclatures for the coding and noncoding variants are described in accordance with the Human Genome Variation Society Nomenclature standards. The promoter variants are described in this study by considering the transcription site to be the +1 nucleotide.27
ResultsFive different Jordanian families with nephropathic cystinosis from different geographical locations in Jordan and one family from Sudan (Family 5) were investigated in this study for the CTNS mutation. Consanguinity was reported for all six of the families. All of the probands initially presented with Fanconi syndrome and rickets. Cystinosis was suggested by the presence of crystals in the cornea upon ophthalmological evaluation by age 2 years in most of the probands. The 57-kb deletion was not found in all of the families and that was evidenced by the presence of the 250-bp PCR product using the LDM2/exon 4 primers and the 266-bp PCR product by using the LDM1/D17S829 primers (data not shown). Amplification of the coding exons (3–12) for the CTNS gene followed by bi-directional DNA sequencing identified three polymorphisms, −294C>T, −180T>C and −118C>T in the CTNS promoter region, one silent (c.504G>A; p.Thr168Thr), one missense (c.779C>T; p.Thr26Ile) variants and two CTNS mutations: c.829dupA (Fig. 1A) and c.890G>A (Fig. 1B). The proband of family 2 in this study was compound heterozygous for both mutations, where c.829dupA was transmitted from the father and c.890G>A was contributed by the mother (Table 1). Both of the parents in family 6 were carriers of the c.829dupA mutation, and their affected children who were not available for testing were presumably homozygous for the same mutation. Both of the probands of Jordanian family 1 and Sudanese family 5 were homozygous for the c.890G>A mutation (Table 1). The C allele encoding Thr amino acid of the c.779C>T variant was in cis with the mutant alleles for both c.829dupA and c.890G>A mutations in the Jordanian families, whereas in the Sudanese proband, the T allele coding for Ile amino acid was in cis with mutant A allele for the c.890G>A mutation.
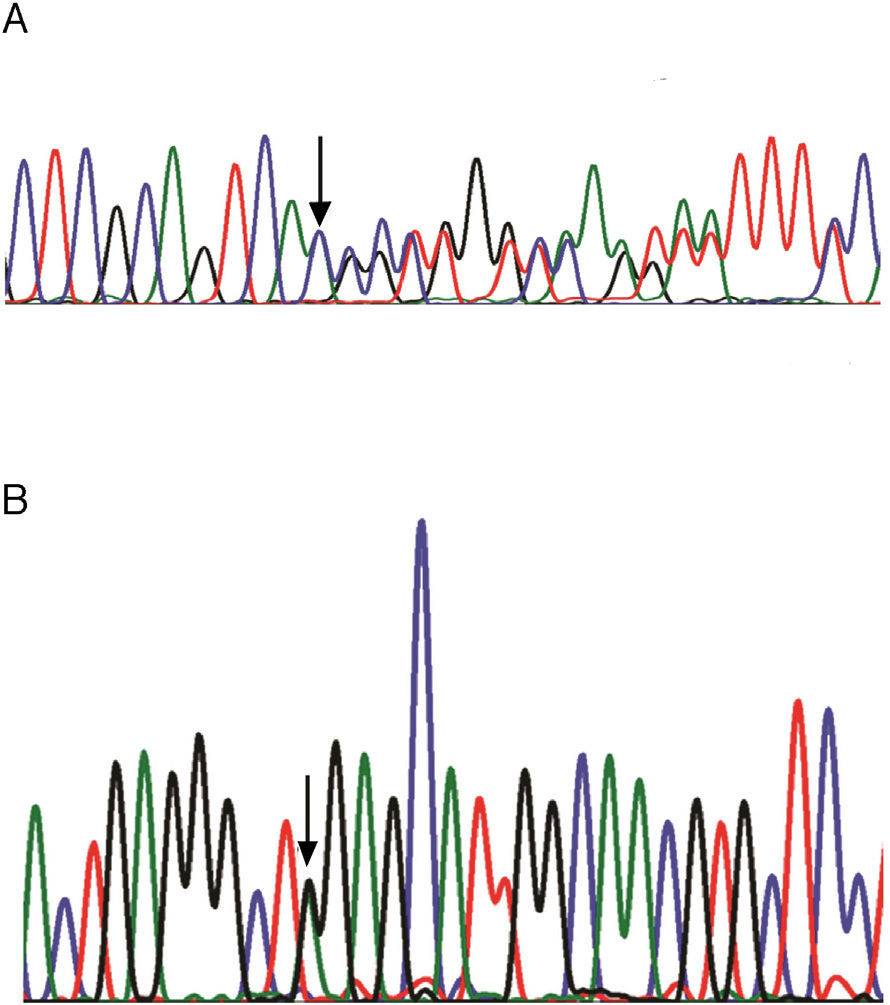
(A) DNA sequence chromatograms for heterozygous genotype (mother, family 3) of the c.829dupA mutation in exon 10 of the CTNS gene. (B) DNA sequence chromatograms for heterozygous genotype (mother, family 1) of the c.890G>A mutation in exon 11 of the CTNS gene. The arrows indicate the locations of the mutations.
Mutations in the CTNS gene characterised in Jordanian families.
| Subject | Symptomatology | c.829dupA | c.890G>A |
|---|---|---|---|
| Family 1 | |||
| Proband | Yes | WT/WTa | A/A |
| Father | No | WT/WT | G/A |
| Mother | No | WT/WT | G/A |
| Sister | No | WT/WT | G/A |
| Grandmother | No | WT/WT | G/G |
| Family 2 | |||
| Proband | Yes | WT/829dupA | G/A |
| Father | No | WT/829dupA | G/G |
| Mother | No | WT/WT | G/A |
| Brother | No | WT/WT | G/G |
| Family 3 | |||
| Proband | Yes | 829dupA/829dupA | G/G |
| Father | No | WT/829dupA | G/G |
| Mother | No | WT/829dupA | G/G |
| Brother | No | WT/829dupA | G/G |
| Family 4 | |||
| Proband | Yes | WT/829dupA | G/G |
| Family 5 | |||
| Proband | Yes | WT/WT | A/A |
| Father | No | WT/WT | G/A |
| Mother | No | WT/WT | G/A |
| Family 6 | |||
| Father | No | WT/829dupA | G/G |
| Mother | No | WT/829dupA | G/G |
This is the first molecular study on Jordanian patients with cystinosis infantile nephropathic cystinosis. The 57-kb deletion was not found in any of the Jordanian probands using two different multiplex polymerase chain reactions. Our results agree with previous regional studies in which the 57-kb deletion was not found in Saudis, Egyptians, Emiratis, Turkish or Iranians.4,7–10 The 57-kb deletion was previously found in homozygous genotypes in 48 of 108 (44%) patients of European descendant living in America, and this deletion also accounts for 34% and 65% of the CTNS mutant alleles in patients of Spanish and German origin.18,22,29
In this study, we identified two frameshift mutations in the CTNS gene in five Jordanian families and one family from Sudan with a history of nephropathic cystinosis. The c.829dupA (p.Thr277Asnfs*19) mutation, which is located in exon 10, resulting in an amino acid substitution for residues 277–294 with the exception of the Phe residue at position 282 and a premature stop codon at residue 295 (Glu295X). The c.829dupA mutation was described in a compound heterozygous genotype with the 57-kb deletion in Belgium and was recently found in a homozygous genotype in three probands and one sibling from Egypt.7,30 The second mutation, c.890G>A mutation, which is located in exon 11, converts the Trp 297 TGG codon into a TAG stop codon (p.Trp297X). The c.890G>A was originally described in a heterozygous genotype in a male from South Italy with first-cousin parents; as well as for the c.779C>T variant that was found in 5 Italian patients.15 Both of the mutations in this study were found in the proband of family 2 as a compound heterozygous genotype that was not previously described. The finding of the compound heterozygous genotype is not usually expected in a consanguineous family. However, several examples have been reported in which two different mutations create compound heterozygotes in consanguineous families in Mediterranean populations. For example, Lebanese patients from an extended consanguineous family with Bardet-Biedl syndrome were found to be heterozygous for two missense mutations, p.Ser311Ala and p.Val11Gly, in the BBS10 gene.31 Similarly, in an inbred pedigree in an Israeli Arab family with congenital nephrotic syndrome, two patients carried both p.Gln380X and p.Cys721fs mutations in the NPHS1 gene as a compound heterozygous genotype.32 The case we report is less extreme in the sense that both mutations (p.Thr277Asnfs*19 and p.Glu295X) are seen in homozygous (four Jordanian families) and in heterozygous (one Jordanian family) living in different and distant locations; but founder effects likely account for their joint occurrence. This possibility could be further ascertained by additional genotyping and analyses of haplotype status in the probands and families.
The inheritance of the same c.890A mutant allele in cis with the C allele of the c.779C>T variant in Jordanian and in cis with the T allele in Sudanese patients suggests an independent origin of the same mutation in the Arabic population. The association of the c.779C allele with both of the mutant alleles for c.890A and c.829dupA is an additional DNA marker that can be used for prenatal molecular diagnostics of cystinosis in Jordanian families.
Both the c.829dupA in exon 10 and c.890G>A in exon 11 resulted in a truncated cystinosin protein that, if stable, would lack the 6th and the 7th transmembrane domains and the five-amino-acid ‘GYDQL’ lysosomal sorting signal at the c-terminal tail of the protein. Additionally, the c.829dupA mutation resulted in eight amino acid substitutions within the first nine amino acids of the third cytoplasmic loop, which is composed of 19 amino acids (280–298) between the fifth and sixth potential transmembrane domains.24,25 The deletion of the first nine amino acids of this loop (amino acids 280–288) coupled with the deletion of the defined carboxyl ‘GYDQL’ motif resulted in the complete relocalisation of cystinosin-GFP to the plasma membrane without a signal remaining in the lysosomes.24,25 Therefore, both of the mutations that were identified in this study potentially resulted in the partial redirection of cystinosin to the plasma membrane, and the c.829dupA mutation may also result in the complete relocalisation of cystinosin to the plasma membrane.
The proband of family 4 who was heterozygous for the c.829dupA mutation inherited haplotype −294T/T, −180C/C and −118T/T for the three polymorphisms in the CTNS promoter region spanning −85 to −406 nucleotides relative to the CTNS +1 transcription site. The C-to-T substitution is 294bp upstream of the transcription site of the CTNS gene, which is part of the Sp1 consensus binding (−299 to -293) sequence in the CTNS promoter.24 An in vitro study using a luciferase vector reporter gene construct harbouring the −294T allele that was transfected transiently in human kidney tubular cells (HK2) showed a 30% reduction in luciferase transcriptional activity compared to −294C construct.28 Similarly, a functional study showed an 81% reduction in CAT reporter gene constructs harbouring the −295C mutant allele of the CTNS promoter.24 However, four patients carrying the −295G>C mutation were also carrying two allelic mutations in the coding region for the CTNS gene, which may contradict a role of promoter mutation in the disease.15 In proband 4, the second mutated allele may be within the CTNS intronic regions, which were not sequenced in this study, and this mutation may affect the splicing processes.16
There is evidence for the allelic heterogeneity of nephritic cystinosis disease in Arabic patients, as twenty-three known and new mutations in the CTNS mutation have been characterised in Arabic individuals thus far.4–8 These mutations spread across the CTNS gene, mainly in the coding exon number 3 and exons 6–12, and include several types, including missense, nonsense, frameshift and splicing mutations. Twelve of the 23 mutations were novel.4–8 Four mutations, including the two mutations that were described in this study (Thr277Asnfs*19 and p. Trp297), i.e., p. G308R and p.G339R, were characterised in more than one Arabic nationality and have also been characterised in other parts of the world.4,7,9,15,18,29,30
However, the c.681G>A; p.E227E mutation that substituted for the last base of exon 9 of the CTNS gene and created a new cryptic donor site was previously found exclusively in several patients from Middle Eastern countries but not in Jordan.4,7,9,10 Despite the notable prevalence of the c.681A mutant allele in homozygous and heterozygous genotypes in cystinosis patients in the Middle East, further haplotype analysis for chromosome 17 is required to determine whether this is a founder mutation in the region. The geographical distribution of the c.681G>A allele in several Middle Eastern populations was analysed by Soliman et al., who proposed that the origin of this mutation is in Eastern Iran.7 Similarly, putative founder mutations, such as c.753G>A and c.898-900_24del27, have been characterised in French Canada and in the western province of Brittany, France, respectively.33,34
In summary, we have identified two loss-of-function mutations in the CTNS gene in several Jordanian families and one family from Sudan. These two mutations extend the regional mutation database for the orphan disease cystinosis. Both of the mutations were originally described in heterozygous genotypes in European patients and have been found in Egyptians, Jordanians and Sudanese individuals. These results will facilitate the molecular genetic diagnosis of cystinosis in Jordan.
Conflict of interestThe authors declare no conflict of interest.
We are thankful to the patients and their family members for their participation in the study. This work was supported by a grant (Grant no. 2004.0044) from the Deanship of Research at Jordan University of Science and Technology.



