
We present the case of a 59-year-old woman referred to nephrology in 2009 for CKD (Cr 1.36 mg/dl, MDRD 45 ml/min/1.73 m2).
As background of interest she had been undergoing follow-up by the gastroenterology department since 2008 due to alteration of liver function tests (AST 89 UI/l [0–37]; ALT 141 UI/l [0–40]; ALP 696 UI/l [70–208]; GGT 349 [10–50] BRBt 0.4 mg/100 [0.1–1.2]) without conclusive results regarding the etiology, the ultrasound and liver biopsy did not show pathological findings. Empirical treatment with ursodeoxycholic acid was prescribed with partial response.
We extended the study and she had no albuminuria or alterations in the urinary sediment. We detected ANA 1/40 with negative anti-dsDNA and ENAS antibodies, normal C3/C4. Ultrasound showed small kidneys (9 cm), with cortical hyperechogenicity and poor corticomedullary differentiation, so it was decided not to perform kidney biopsy.
She denied previous ingestion of nephrotoxic drugs: she had never had episodes of urinary tract infections or episodes of kidney stones.
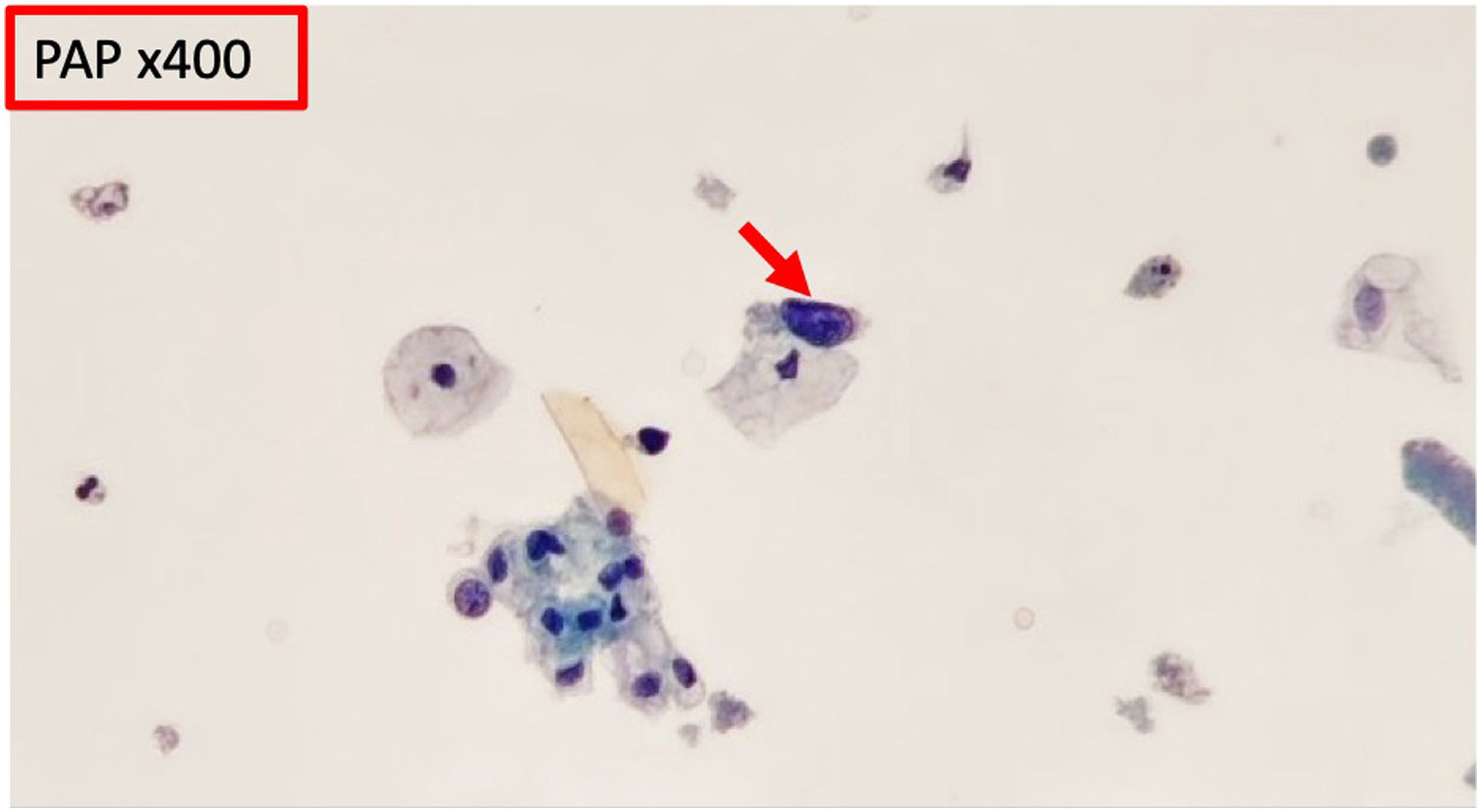
In 2015 microhematuria appeared. Urine cytology was requested and it was described as: “single cells with discrete atypical features, hyperchromatic nuclei, scant cytoplasm” (Fig. 1).
 compared to adjacent normal urothelial cells.")
In 2021 we reinterrogated the family history: consanguineous parents (cousins), the patient’s mother had CKD (stage unknown) and died from acute myocardial infarction (MI) at 68 years of age; patient’s brother also had CKD (stage unknown) and died of MI at the age of 59 year. The relatives of the maternal family had a multiple history of ischemic heart disease.
Therefore, we requested screening for Fabry disease (measuring the enzyme activity of the enzyme α -galactosidase-A and Lyso-globotriaosylceramide levels in dried blood drops), which was negative.
In view of the family profile and an unknown-cause CKD, we decided to request a genetic study of renal diseases, detecting a homozygous pathogenic variant in the FAN1 gene [c.2854C > T p.(Arg952Ter)] associated with karyomegalic interstitial nephritis.
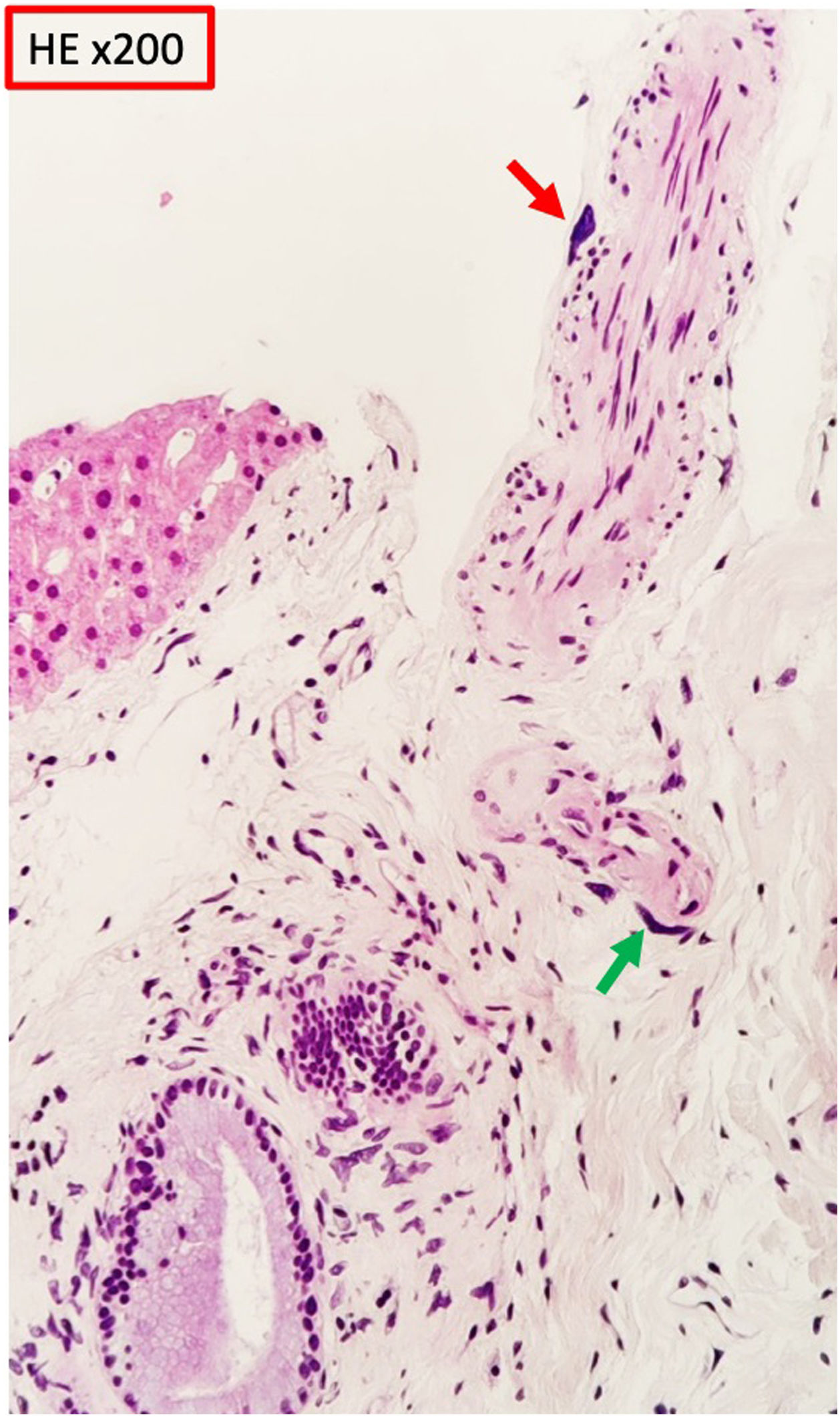
We informed the gastroenterology department, who decided to repeat a liver biopsy, identifying in the connective tissue of a subcapsular portal space cells of karyomegalic appearance, some in perineural and perivascular arrangement and showing large, irregular and markedly hyperchromatic nuclei (Fig. 2). None of the biopsies showed cholestatic damage or liver fibrosis. From the analytical point of view, a partial response to ursodeoxycholic acid persists, but alkaline phosphatase levels remain elevated with fluctuations parallel to renal function.
 and perivascular (green arrow) arrangement showing large, irregular and hyperchromatic nuclei.")
Karyomegalic interstitial nephritis is a rare disease that is transmitted in an autosomal recessive manner. It was first described in 19741 and the term “karyomegalic interstitial nephritis” was subsequently introduced in 1979.2 In 2012 Zhou et al. identified mutations in homozygosity in the FAN1 gene that cause the disease,3 and subsequently other mutations have been discovered in the same gene.4
The FAN1 gene encodes for the protein Fanconi anemia-associated nuclease 1, involved in DNA repair. This protein is expressed in the parenchyma of multiple tissues (liver, neuronal tissue, myocardium, thyroid and endothelial cells), with the highest expression in the kidney.5
Pathogenic variants of this gene have been associated with interstitial nephritis and lesions in other organs, for example, in the liver. The biopsies show cells characterized by large, irregular, hyperchromatic nuclei. These cells have also been described in urine cytology of affected patients.
There are fewer than 50 published cases of this disease. In general, they describe the diagnosis of CKD in patients from the third decade onwards, with variable intra- and inter-familial progression, reaching terminal CKD.5,6
The diagnosis of certainty is the finding of a pathogenic variant of the FAN 1 gene in homozygosis, since the inheritance pattern is autosomal recessive.
In our patient the result of the genetic study and the finding of characteristic cells in urine and in the liver biopsy confirm the diagnosis of this disease, which explains her renal and previously unidentified hepatic involvement.
FinancingThis work has not received any funding.



