




La hipertensión arterial maligna puede presentarse en forma de insuficiencia renal grave, en ocasiones acompañada de microangiopatía trombótica, que se manifiesta en forma de anemia hemolítica no inmune y trombocitopenia. En estos casos, inicialmente puede resultar difícil diferenciarla de otros procesos con características similares de presentación, fundamentalmente la púrpura trombótica trombocitopénica y el síndrome hemolítico urémico atípico. El manejo de estos tres cuadros difiere sustancialmente, por lo que es fundamental disponer de algunas claves clínicas precoces para realizar el diagnóstico diferencial. Presentamos el caso de un paciente que debutó con hipertensión arterial maligna, microangiopatía trombótica e insuficiencia renal grave. Realizamos una revisión sobre todos los aspectos mencionados.
CASO CLÍNICO
Paciente de 40 años, pakistaní. Temporero en la agricultura. Fumador de larga evolución. Sin revisiones ni medicación habitual.
Un mes antes del ingreso acudió al centro de salud por cefalea y malestar general, detectándose hipertensión arterial (HTA) grado III (220/115 mmHg), y se inició un hipotensor. El cumplimiento no fue estricto. Posteriormente, 10 días antes del ingreso inicia cuadro de epigastralgia, náuseas y disminución de la ingesta. No refería cefalea, ni otra sintomatología. Ante la persistencia de la epigastralgia consultó en urgencias de nuestro hospital.
Exploración física: presión arterial (PA) 243/132 mmHg; auscultación cardíaca, tonos rítmicos con soplo sistólico aórtico; auscultación pulmonar, normoventilación; abdomen blando, depresible, doloroso en epigastrio, signo del rebote negativo, peristaltismo conservado; extremidades inferiores, no edemas ni signos de trombosis venosa; no signos de focalidad neurológica; en el fondo de ojo presentaba retinopatía hipertensiva grado IV en ambos ojos, con hemorragias y exudados en 360º de retina, edema perivascular, hemorragias peripapilares, papiledema y estrella macular (figura 1); electrocardiograma con signos de hipertrofia ventricular con sobrecarga sistólica; radiografía de tórax, cardiomegalia sin signos de descompensación.
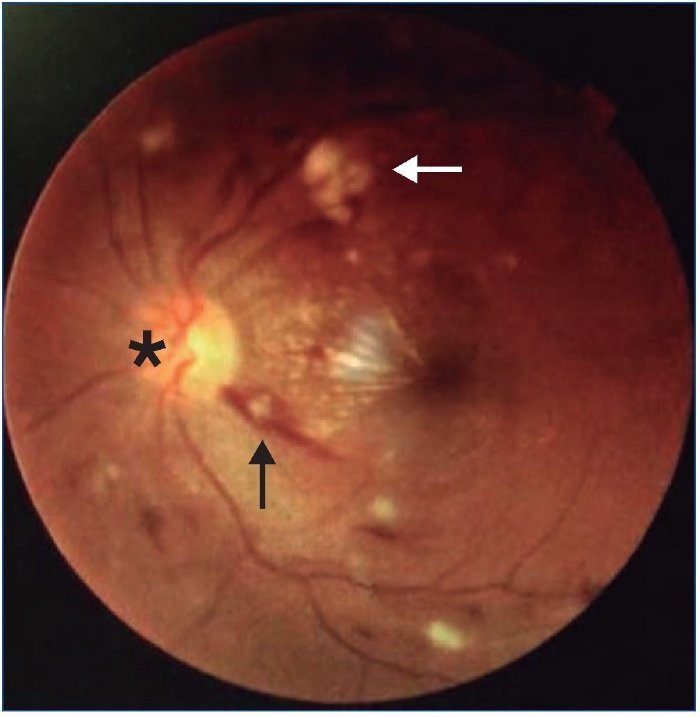
Figura 1. Fondo de ojo realizado al ingreso que muestra hemorragias (flecha negra), exudados algodonosos (flecha blanca) y edema de papila (asterisco).
Analítica de urgencias: destacaba una insuficiencia renal grave (creatinina sérica 7,9 mg/dl). Otros parámetros: Na 135 mEq/l; K 3,99 mEq/l; Cl 92,2 mEq/l; pH 7,47; pCO2 35 mmHg; HCO3Na 23,3 mmol/l. En el hemograma destacaba una anemia y plaquetopenia con: hemoglobina 9,9 g/dl; leucocitos 6.500/µl; plaquetas 70.000/µl. La hemostasia era normal. En el frotis de sangre periférica no se detectaban esquistocitos. Sedimento urinario 1-5 hematíes/campo; proteinuria +++ en tira reactiva. Ecografía: riñones de tamaño normal, simétricos, con aumento de la ecogenicidad sin apreciar dilatación del sistema colector.
Se trató con perfusión de solinitrina intravenosa durante 12 h, con mejoría parcial de las cifras tensionales, y posteriormente se iniciaron hipotensores orales (diurético del asa, antagonista del calcio, antagonista de los receptores de la angiotensina II, bloqueador alfa), con progresiva mejoría tensional.
El tercer día, la creatinina sérica alcanzó el pico de 9,3 mg/dl, a pesar de diuresis adecuada. Por ello se inició hemodiálisis y, no pudiendo descartar una enfermedad glomerular primaria, se administraron tres bolos de 500 mg de metilprednisolona en días sucesivos, con pauta oral posterior a dosis de 80 mg. No se indicó realizar una biopsia renal ante el imperfecto control tensional y la persistencia de la trombopenia.
Un segundo frotis realizado el cuarto día del ingreso detectó esquistocitos en sangre periférica, junto con test de Coombs directo negativo, lactatodeshidrogenasa (LDH) 497 U/l (valores normales [vn]: 135-225 U/l) y haptoglobina < 7,44 mg/dl (vn: 50-150), todo ello sugestivo de anemia hemolítica de origen microangiopático. Se realizó extracción para la determinación de ADAMTS13 (A Desintegrin and Metalloproteninasa with ThromboSpondin type 1 motif, member 13), que se envió a un laboratorio externo.
Otros parámetros analíticos séricos fueron: proteínas totales 6,7 g/dl; albúmina 3,55 g/dl; colesterol 164 mg/dl; calcio 8,94 mg/dl; fósforo 8,19 mg/dl; GOT 18 U/l; GPT 21 U/l; FA 89 U/l; GGT 19 U/l; bilirrubina 0,43 mg/dl; PTH 453 pg/ml (vn: 14-72); vitamina D 11,75 ng/ml (vn: 30-150); TSH 0,74 µU/ml (vn: 0,55-4,78); cortisol basal 18,8 µg/dl; vitamina B12 139 pg/ml (vn: 160-450); ácido fólico 7,05 ng/ml (vn: 3,1-19,9). B-2-microglobulina 20,5 µg/ml (vn: 1,09-2,53). Serologías de VHB, VHC y VIH negativas. En orina la proteinuria era 1,32 g/24 h. La inmunofijación no detectó paraproteínas monoclonales.
El estudio de autoinmunidad fue: IgG 1.160 mg/dl (vn: 768-1.632); IgM 80,2 mg/dl (vn: 60-263); IgA 275 mg/dl (vn: 68-378); IgE 607 mg/dl (vn: 0-180); C 3 86,7 mg/dl (vn: 79-152); C 4 23,8 mg/dl (vn: 10-40); factor B 31,4 mg/dl (vn: 10-40); proteína C reactiva 0,38 mg/dl (vn: 0,02-0,61 ); ASO 34,8 UI/ml (vn: 0-330); DNASA B < 50 UI/ml (vn: 0-200); anticuerpos frente a membrana basal negativo; C-ancas negativo; P-ancas negativo; ANA negativo; anti-ADN bicatenario negativo; factor reumatoide < 20 UI/ml (vn: 0-20); anticuerpos anticardiolipina negativos; anticuerpos antitiroideos normales.
Se inició estudio de HTA secundaria. La determinación de tóxicos incluyendo cocaína fue negativa. Las catecolaminas y metanefrinas en orina eran normales. Los valores de renina y aldosterona no estaban alterados. Una arteriografía renal resultó normal, descartando la HTA vasculorrenal. En el ecocardiograma presentaba patrón de hipertrofia concéntrica ventricular izquierda, con llenado restrictivo y con grosor del septo y la pared posterior de 15 mm.
El tratamiento hipotensor consiguió controlar óptimamente la PA. De modo paralelo se normalizó la cifra de plaquetas y desaparecieron los signos de hemólisis. Tras ello se practicó una biopsia renal percutánea, que evidenció como signo más llamativo una afectación difusa y grave de las arteriolas renales, mostrando lesiones de endarteritis proliferativa (figura 2) y estrechamiento de las luces junto con lesiones de necrosis fibrinoide en la pared (figura 3). Los glomérulos presentaban cambios isquémicos sin presencia de trombos en la luz de los capilares. Se detectaban signos de discreta nefritis tubulointersticial. La inmunofluorescencia era negativa.
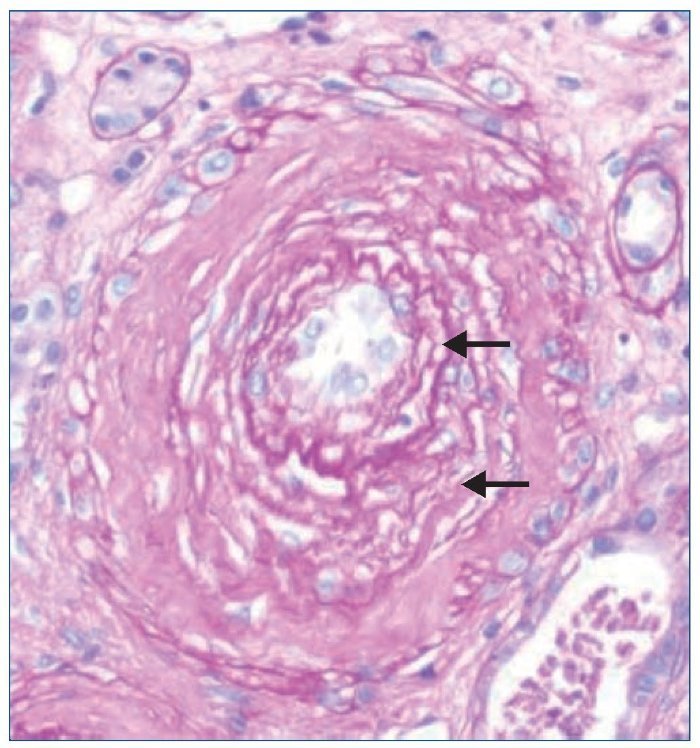
Figura 2. Fibrosis subintimal de la pared arteriolar con desdoblamiento de las fibras elásticas (flechas). Tinción de PAS (magnificación ×10).
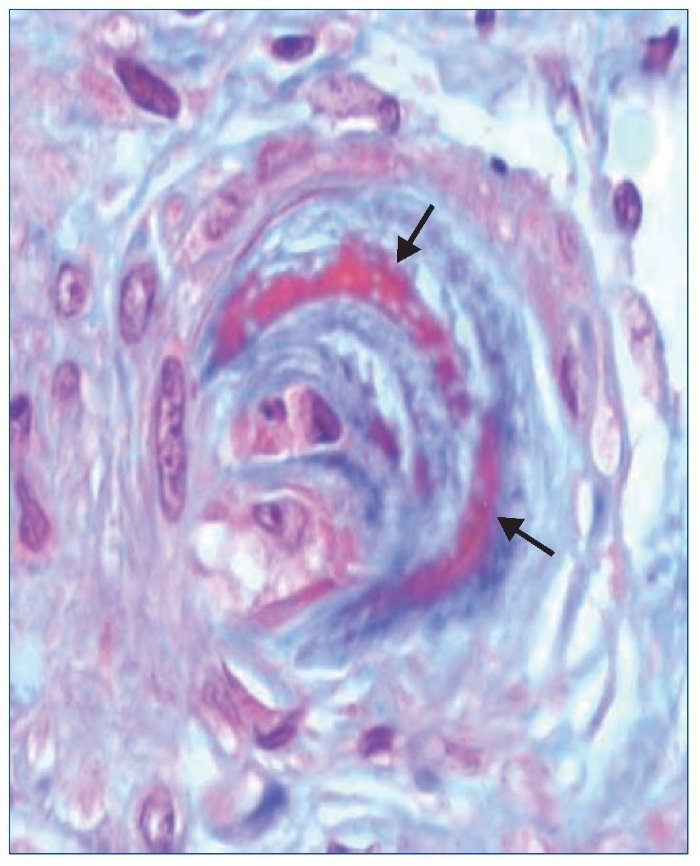
Figura 3. Arteriola renal con lesiones de necrosis fibrinoide en la pared (flechas). Tinción de tricrómico de Masson (magnificación ×20).
Se recibió el resultado del ADAMTS13, que fue normal, descartándose la púrpura trombótica trombocitopénica (PTT). Respecto al síndrome hemolítico urémico atípico (SHUa), no se solicitó un estudio genético ante la normalidad de los valores séricos del complemento y las características clínicas y evolutivas del cuadro, que nos llevaron a descartar esta etiología por exclusión.
En la evolución posterior suspendimos los corticoides y ante la ausencia de recuperación de la función renal el paciente fue incluido definitivamente en programa de hemodiálisis. Un segundo fondo de ojo realizado mes y medio después del primero mostró la completa reversibilidad de las lesiones de retinopatía hipertensiva.
Los diagnósticos finales fueron los de HTA de origen esencial malignizada, que cursó con microangiopatía trombótica (MAT) e insuficiencia renal terminal secundaria a la grave afectación vascular intrarrenal.
DISCUSIÓN
El caso aquí presentado ilustra la dificultad inicial para diferenciar los casos que se presentan con HTA maligna que cursa con MAT —circunstancia que fue descrita por primera vez en 19541 y que según la bibliografía se produce en un 20-40 % de los casos2-4— de las MAT primarias, entre las que destacan por su trascendencia el SHUa secundario a alteraciones en los componentes del complemento y la PTT5-7. A destacar que se ha descrito que estas dos enfermedades pueden presentarse con HTA maligna8-11. Para diferenciar las tres entidades se debe recurrir a datos clínicos o analíticos, pero algunos de estos pueden demorarse en su resultado, como el ADAMTS13, necesario para descartar una PTT, que en la mayoría de centros como el nuestro debe remitirse a un laboratorio externo y esperar varios días para conocer el resultado. En el caso del SHUa, los resultados de los estudios genéticos se pueden demorar mucho más, por lo que en la práctica el diagnóstico inicial suele ser de exclusión. No obstante hay que mencionar que se ha descrito que en algunas series hasta un 14 % de casos catalogados inicialmente como HTA maligna presentaban una alteración hereditaria del complemento12. Cabe preguntarse si este bajo porcentaje justifica la realización del estudio genético en todos los casos, como el aquí presentado.
Las implicaciones prácticas son importantes. Si se sospecha una PTT está indicado el inicio de sesiones de plasmaféresis, procedimiento que no es inocuo13. En caso de sospechar un SHUa estaría indicado iniciar tratamiento con eculizimab, fármaco que posiblemente haya que mantener de por vida, cuyos protocolos de administración todavía no están bien definidos y que hoy por hoy es una terapia muy onerosa14. Si finalmente el diagnóstico se decanta por una HTA maligna, cuyo origen obviamente habría que dilucidar, el tratamiento indicado es el del control de las cifras tensionales. Trabajos previos han mostrado cómo de modo paralelo al control tensional se normalizan los signos de MAT3, hecho que se produjo también en nuestro caso. En una publicación reciente se muestra cómo el grado de incertidumbre inicial llevó a los clínicos a indicar plasmaféresis hasta en un tercio de los casos de pacientes que finalmente eran portadores de una HTA maligna con MAT, pero en los que de inicio no se podía descartar fehacientemente una PTT4. En este trabajo se analiza una serie de 19 casos bien documentados. Los síntomas más frecuentes de presentación fueron neurológicos o gastrointestinales como náuseas/vómitos (42 %) o dolor abdominal. Todos los pacientes presentaban datos de insuficiencia renal grave (creatinina media 5,2 mg/dl). No obstante, la trombopenia era relativamente modesta (media de recuento plaquetario 60.000/ µl). Tras alcanzar el control de la HTA, la mayoría mostró una mejoría de la sintomatología de presentación (100 %) y de la cifra de plaquetas (84 %). No obstante, solo en el 58 % de los casos se objetivó una mejoría significativa de la función renal. Más de la mitad de los pacientes (53 %) precisaron hemodiálisis4. Este perfil de pacientes descrito coincide con el de nuestro caso, en el que llama la atención que el motivo de consulta fue únicamente una epigastralgia.
Para orientarnos en el diagnóstico diferencial, algunos autores han descrito una serie de datos clínicos y analíticos que pueden ayudar a diferenciar estas entidades4,15,16 (tabla 1). Cabe destacar la importancia de la realización precoz de un fondo de ojo17,18. No obstante, ya se ha comentado que se han descrito algunos casos esporádicos de SHUa y PTT que debutaron con HTA maligna acompañante8-11. En nuestro paciente existía una barrera idiomática que dificultaba de modo notorio la anamnesis. Por ello, entre las primeras pruebas realizadas, el fondo de ojo y el ECG cobraron especial importancia a la hora de atribuir a la HTA la presumible etiología de la MAT.

En nuestro caso, dada la incertidumbre diagnóstica inicial, se instauró tratamiento con esteroides ante la posibilidad de una enfermedad glomerular primaria. Entre ellas se ha descrito la nefropatía IgA que debuta con HTA maligna19. Con los resultados de la biopsia se suspendieron los esteroides.
En las series clásicas sobre HTA maligna, la HTA esencial no era la principal etiología. En la revisión de Kincaid-Smith de 1991 representaba el 20 % de los casos20. No obstante, en series más contemporáneas, como la del grupo español del Hospital 12 de Octubre, la HTA esencial representa sin duda la primera causa, y abarca hasta el 75 % de los pacientes17. Por lo tanto, aunque en casos como el nuestro está indicado descartar causas de HTA secundaria21, no debe resultar extraño que los resultados sean negativos. Diversos autores señalan que el principal factor desencadenante de la malignización de la HTA esencial es el mal cumplimiento o el abandono del tratamiento17,22.
En nuestro paciente, los hallazgos histológicos de la biopsia renal eran los previamente descritos en casos similares22-25. Mostraron lesiones vasculares graves secundarias a la HTA como responsables del daño renal. En el riñón, las lesiones más características son la necrosis fibrinoide y la endarteritis proliferativa. La necrosis fibrinoide es posiblemente el resultado de la lesión endotelial por la propia hipertensión, con el depósito de proteínas plasmáticas (fibrina) en el subendotelio. Esta lesión se reconoce como eosinofílica con detritus de células musculares necróticas y restos proteicos en la pared arteriolar. La endarteritis proliferativa (hiperplasia o proliferación miointimal) se caracteriza por un engrosamiento de la íntima constituido por células musculares lisas, restos de membrana basal y mucopolisacáridos, que conlleva un importante estrechamiento de la luz vascular con el consiguiente aumento de la resistencia al flujo de sangre e isquemia parenquimatosa26. En algunos casos puede observarse proliferación en capas de cebolla. En el modelo animal, la lesión más característica es la necrosis fibrinoide, mientras que en el hombre esta lesión se observa raramente, quizá debido a que la presencia de sintomatología condiciona la instauración de tratamiento antes de desarrollarse la necrosis de la pared vascular. La lesión que con más frecuencia se observa en el riñón humano es una arteriosclerosis hiperplásica y una esclerosis glomerular isquémica. Algunos autores han reportado la presencia adicional de microtrombos de fibrina intraglomerulares, que en nuestro caso, al igual que en otras series27, no se detectaron.
Respecto a la fisiopatología de la HTA maligna, puede decirse que la suma de factores genéticos28 y ambientales conduce a un grado crítico de HTA, sobre el que inciden factores locales y sistémicos, reflejo de un desequilibrio entre los mecanismos vasoconstrictores y vasodilatadores, y un incremento de la actividad simpática y del sistema renina-angiotensina-aldosterona en íntima asociación con la disfunción endotelial17. Todos estos mecanismos dan lugar a una lesión de la pared vascular que permite el paso al endotelio de factores plasmáticos (necrosis fibrinoide) que causan un estrechamiento y una obliteración de la pared vascular. El cuadro clínico se caracteriza por una afectación multiorgánica y sus manifestaciones dependen, entre otras, de su repercusión cardíaca, cerebral y renal. No está claro por qué unos pacientes cursan con MAT y otros no. A este respecto se ha sugerido la participación del sistema renina-angiotensina-aldosterona. En un estudio, al comparar 2 grupos de pacientes, unos con HTA grave y otros con HTA maligna, estos últimos presentaban mayores valores de actividad de renina plasmática (ARP) y de aldosterona. Además existía una asociación lineal positiva entre ARP y valores de LDH, marcador de hemólisis. Lo mismo sucedía con la creatinina29. Por ello, este y otros autores justifican el empleo de hipotensores del grupo de los IECA (inhibidores de la enzima de conversión de la angiotensina)/ARA II (antagonistas del receptor de la angiotensina II) en estos pacientes con el objeto de contrarrestar esta activación17,29. El hecho de que en nuestro paciente ya se hubiera iniciado tratamiento con ARA II cuando se tomó la muestra de renina y aldosterona, quizá influyó en que los resultados de estos parámetros no estuvieran elevados. En la serie de Akimoto et al3, 7 de 16 pacientes presentaban MAT. La etiología fue HTA esencial en el 56 %. Estos autores señalan que sus observaciones les llevan a considerar que la vasculatura de los pacientes con MAT puede ser más vulnerable que la de los pacientes sin MAT respecto al efecto de la HTA incontrolada. La vasculatura puede estar afectada por el daño endotelial inducido por la aldosterona elevada. Las diferentes etiologías de la HTA pueden determinar también la susceptibilidad endotelial al incremento de la PA. En otro estudio que revisó 97 casos de HTA maligna, 26 de ellos presentaban anemia hemolítica microangiopática (AHMA). Los autores señalan que la presencia de AHMA en pacientes con HTA maligna es un importante factor predictor, tanto de la insuficiencia renal como de su recuperación2.
Respecto al pronóstico de la HTA maligna, existe una serie de factores descritos que influyen en la supervivencia renal, pues hoy en día, contrariamente a lo que sucedía antaño, con el advenimiento de los modernos tratamientos hipotensores la supervivencia del paciente no suele estar comprometida. Estos factores, entre los que cabe mencionar el grado de insuficiencia renal y el grado de control de la PA, pueden estar presentes en el momento del diagnóstico o a lo largo de la evolución16. A destacar la importancia pronóstica de la proteinuria, puesta de relieve por el grupo español del Hospital 12 de Octubre30.
En resumen, presentamos un caso que ejemplifica la dificultad para diferenciar inicialmente la HTA maligna asociada a MAT de las otras MAT primarias como la PTT y el SHUa, con presentación clínica similar. Para el diagnóstico diferencial y la toma de decisiones terapéuticas precoces existen una serie de datos clínicos y analíticos (tabla 1), que pueden ayudar a los especialistas encargados de estos pacientes a orientar los casos desde el punto de vista práctico.
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.
Correspondencia: José María Peña Porta
Servicio de Nefrología.
Hospital Clínico Universitario Lozano Blesa.
Avenida San Juan Bosco, 15. 50009 Zaragoza.
jpenaporta@gmail.com


