




Autosomal dominant tubulointerstitial kidney disease (ADTKD) comprises a heterogeneous group of rare hereditary kidney diseases characterized by family history of progressive chronic kidney disease (CKD) with bland urine sediment, absence of significant proteinuria and normal or small-sized kidneys. Current diagnostic criteria require identification of a pathogenic variant in one of five genes – UMOD, MUC1, REN, HNF1β, SEC61A1. The most prevalent form of ADTKD is uromodulin-associated kidney disease (ADTKD-UMOD).
Genetic study of a Portuguese family diagnosed with familial juvenile hyperuricemic nephropathy (FJHN), one of the nosological entities in the spectrum of ADTKD, revealed a previously unreported large deletion in UMOD encompassing the entire terminal exon, which strictly cosegregated with CKD and hyperuricemia/gout, establishing the primary diagnosis of ADTKD-UMOD; as well as an ultra-rare nonsense SLC8A1 variant cosegregating with the UMOD deletion in patients that consistently exhibited an earlier onset of clinical manifestations.
Since the terminal exon of UMOD does not encode for any of the critical structural domains or amino acid residues of mature uromodulin, the molecular mechanisms underlying the pathogenicity of its deletion are unclear and require further research.
The association of the SLC8A1 locus with FJHN was first indicated by the results of a genome-wide linkage analysis in several multiplex families, but those data have not been subsequently confirmed. Our findings in this family revive that hypothesis.
La enfermedad renal tubulointersticial autosómica dominante (ADTKD) comprende un grupo heterogéneo de enfermedades renales hereditarias raras caracterizadas por historia familiar de enfermedad renal crónica (ERC) progresiva con sedimento urinario blando, ausencia de proteinuria significativa y riñones normales o de tamaño pequeño. Los criterios de diagnóstico actuales requieren la identificación de una variante patogénica en uno de cinco genes: UMOD, MUC1, REN, HNF1β, SEC61A1. La forma más frecuente de ADTKD es la enfermedad renal asociada a uromodulina (ADTKD-UMOD).
El estudio genético de una familia portuguesa diagnosticada con nefropatía hiperuricémica juvenil familiar (FJHN), una de las entidades nosológicas en el espectro de ADTKD, reveló una deleción extensa en el gen UMOD abarcando todo el exón terminal, no reportada anteriormente, que cosegregaba estrictamente con ERC e hiperuricemia/gota, permitiendo establecer el diagnóstico de ADTKD-UMOD; así como una variante sin sentido, ultra rara, en el gen SLC8A1 que cosegregaba en pacientes que consistentemente exhibieron un inicio más temprano de la manifestaciones clínicas.
Dado que el exón terminal de UMOD no codifica ninguno de los dominios estructurales críticos o de los aminoácidos de la uromodulina madura, los mecanismos moleculares subyacentes a la patogenicidad de su eliminación no están claros, requiriendo más investigación.
La asociación del locus SLC8A1 con la FJHN fue indicada por primera vez por los resultados de un genoma en varias familias multiplexadas, pero esos datos no se han confirmado posteriormente. Nuestros hallazgos en esta familia reavivan esa hipótesis.
The term autosomal dominant tubulointerstitial kidney disease (ADTKD) refers to a heterogeneous group of rare hereditary kidney disorders characterized by tubular damage and interstitial fibrosis, in the absence of glomerular lesions.1–3 Progressive chronic kidney disease (CKD) with bland urine sediment, absence of significant proteinuria, and normal- or small-sized kidneys are the typical clinical features, with a significant proportion of patients also developing hyperuricemia and gout at young ages. The nosology of ADTKD has recently been revised into a gene-based classification system,1 also encompassing the entities formerly designated “medullary cystic kidney disease (MCKD)” and “familial juvenile hyperuricemic nephropathy (FJHN)”. As a group, ADTKD constitutes the third most common genetic disorder in adults with CKD, after autosomal dominant polycystic disease and the collagen type IV-related kidney diseases.2,3 Histopathological findings are nonspecific and definitive diagnosis of ADTKD requires identification of a pathogenic variant in one of the underlying genes1 – i.e. those encoding uromodulin (UMOD), also known as uromucoid or Tamm-Horsfall protein; mucin 1 (MUC1); hepatocyte nuclear factor 1β (HNF1β); renin (REN); or the α1 subunit of SEC61 translocon (SEC61A1).2,3 The most prevalent form of ADTKD is ADTKD-UMOD, which is typically associated with early-onset hyperuricemia and gout.2–4 More than 225 deleterious UMOD variants are reported at The Human Gene Mutation Database (https://www.hgmd.cf.ac.uk/ac/gene.php?gene=UMOD; accessed on 15.08.23), of which the large majority (95.3%) are missense single base-pair substitutions, most of them clustered in exons 3 and 4.3–6
A possibly distinct form of ADTKD – designated FJHN type 3 (FJHN3) – has been tentatively mapped to chromosome 2p22.1-p21,7 by genome-wide linkage analysis in 5 multiplex families with ADTKD. The most likely candidate in that genomic interval was the gene encoding solute carrier family 8 member 1 (SLC8A1), a sodium/calcium exchanger,8 but no pathogenic SLC8A1 sequence variants could be identified in any of the probands7 – however, with the sequencing methods used to that end, copy number variants (CNVs) might have been missed. So far, FJHN3 remains classified as a “phenotype description or locus, molecular basis unknown” in the Online Mendelian Inheritance in Man® catalog (OMIM®; https://www.omim.org/entry/614227; accessed on 15.08.23) and no SLC8A1 variants have ever been established to cause ADTKD.
We report herein the genotype–phenotype correlation analysis of a Portuguese family with ADTKD/FJHN that suggest a possible novel genetic mechanism underlying uromodulin-associated kidney disease and provide evidence supporting a pathogenic role for SLC8A1 variants in the development and natural history of ADTKD.
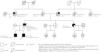
Clinical reportThe proband (IV:2; Fig. 1) was a 27-year-old white Portuguese male, referred to the nephrology clinic for evaluation and treatment of CKD. His medical history was relevant for nocturnal enuresis until age 10; arterial hypertension diagnosed at age 19, well controlled ever since with low dose lisinopril (2.5mg, QD); and hyperuricemia. On the baseline laboratory workup, the estimated glomerular filtration rate was 49ml/min/1.73m2 (CKD-Epidemiology Collaboration creatinine equation, version 2009), the serum uric acid level (831μmol/L) was disproportionately increased to kidney function, and the urinalysis was unremarkable. Magnetic resonance imaging showed normal-sized, normal-shaped kidneys containing multiple small cysts in the medulla and at the corticomedullary junction. Despite continued therapy with allopurinol, the patient suffered several gout attacks during clinical follow-up.

The pedigree chart (Fig. 1) illustrates the unequivocal autosomal dominant pattern of inheritance of the hyperuricemic nephropathy and CKD in this family. The typical clinical phenotype was characterized by (i) hypertension, hyperuricemia and gout as presenting manifestations; (ii) progressive CKD diagnosed in the second to the fourth decades of life; (iii) absence of proteinuria or hematuria; and (iv) normal-sized kidneys with medullary cysts seen on imaging studies.
Genetic diagnosis workupGenetic testing using a customized next-generation sequencing (NGS)-based diagnostic gene panel targeted to ADTKD/MCKD/FJHN (Ipatimup Diagnósticos, i3S – Instituto de Investigação e Inovação em Saúde; University of Porto, Portugal), which includes the SLC8A1 gene, revealed that both the proband and his affected older brother (pedigree subject IV:1) were heterozygous for an ultra-rare nonsense SLC8A1 allele – c.16C>T p.(Arg6*) (Table 1 and Fig. 2A) –, classified as “variant of uncertain significance (VUS)”, according to standard genetic nomenclature guidelines.9 However, since the SLC8A1 p.(Arg6*) allele was not detected in their affected paternal aunt (pedigree subject III:5), it could not explain the segregation of kidney disease in the family.
Genomic identifiers and population prevalence of the UMOD, SLC8A1 and PKD1 variants identified in this family.
| Gene name | RefSeq accessiona | Genomic/coding DNA change | Protein change (predicted) | dbSNP-identifierb | gnomADc population frequency data: allele count/total allele number (sequence type) |
|---|---|---|---|---|---|
| UMOD | NC_000016.10 | g.(20335481_20333375)_(20333051_?)del | ? | – | Novel |
| SLC8A1 | NM_021097.5d | c.16C>T | p.(Arg6*) | rs767111343f | 1/152076 alleles (genomes)h |
| PKD1 | NM_001009944.3e | c.9170T>C | p.(Val3057Ala) | rs758627119g | 1/160674 alleles (exomes)i |

Sanger sequencing demonstrating (A, upper left) the presence of the variant SLC8A1 c.16C>T p.(Arg6*) in the affected brothers (IV:1 and IV:2) and its absence in the affected paternal aunt (III:5), the healthy uncle (III:4) an the mother (III:1); and (A, upper right) the presence of the variant PKD1 c.9170T>C p.(Val3057Ala) in the two affected brothers (IV:1 and IV:2) and their healthy mother (III:1). (B) Quantitative polymerase chain reaction (qPCR) results confirmed the heterozygous UMOD deletion involving exon 11 in all affected family members (IV:1, IV:2, III:5) and its absence in the unaffected ones (III:4, III:1).
Coincidentally, the proband's older brother underwent further genetic testing abroad (Hospices Civils de Lyon; Bron, France), where the use of an extended kidney-disease gene panel allowed to identify (i) a previously unreported large terminal deletion in the UMOD gene, encompassing the entire exon 11, classified as “likely pathogenic” (Table 1); and (ii) an ultra-rare missense variant in the gene encoding polycystin-1 (PKD1) – c.9170T>C p.(Val3057Ala) (Table 1) –, classified as VUS. Since the latter was inherited by the two affected brothers (pedigree subjects IV:1 and IV:2; Fig. 2A) from their healthy mother (pedigree subject III:1; Fig. 2A), who exhibited no kidney cysts on ultrasonography examination at age 63, its pathogenicity could be confidently excluded.10
The UMOD single-exon deletion had been overlooked in the proband's original genetic testing, because the bioinformatic algorithm to detect CNVs using the NGS panel data11 had not yet been validated by the laboratory. Using a quantitative polymerase chain reaction (qPCR) approach, heterozygosity for the UMOD exon 11 deletion was subsequently demonstrated in the other two affected family members (pedigree subjects IV:2 and III:5; Fig. 2B), while it was excluded in the healthy relatives available for genotyping (pedigree subjects III:1 and III:4; Fig. 2B).
Notably, the deceased father of the two affected brothers (pedigree subject III:2), who was an obligate carrier of both the UMOD exon 11 deletion and the SLC8A1 p.(Arg6*) allele, needed kidney replacement therapy (KRT) at age 43, while his also affected younger sister (pedigree subject III:5) did so only at age 55. The earlier clinical presentations of ADTKD in the double heterozygous individuals (pedigree subjects III-2, IV-1 and IV-2), as compared to the significantly later disease onset observed in their relative who inherited only the UMOD variant (III-5), suggest that heterozygosity for the nonsense SLC8A1 p.(Arg6*) allele had a pathogenic or disease modifier role, which is in accordance with its predicted protein truncating effect.9,12
DiscussionThis study illustrates some of the major challenges that clinicians and molecular geneticists encounter in the diagnostic approach to hereditary kidney disorders and highlights the importance of detailed clinical assessment of affected individuals and the potential usefulness of family segregation analysis for reclassification of VUS.13
In families with ADTKD, the specific genetic diagnosis has important implications for the management, prognosis, genetic counseling, prevention of recurrence, and screening of individuals at risk.1 In our family, coincidental genetic testing of two brothers clinically diagnosed with ADTKD, independently performed at different laboratories using distinct gene panels for kidney diseases, yielded three major results: (i) identification of a novel single-exon terminal deletion in UMOD that strictly cosegregated with kidney disease and gout, which constitutes strong clinical evidence of pathogenicity; (ii) detection of an ultra-rare SLC8A1 nonsense allele that segregated with a more severe ADTKD phenotype, which, together with the predicted protein truncating effect, suggest pathogenicity9,12; (iii) detection of an ultra-rare PKD1 missense allele that did not segregate with polycystic kidneys, which is in agreement with the classification of neutral polymorphism attributed to the more common p.(Val3057Met) variant affecting the same codon.14
Uromodulin is a kidney-specific protein, mainly anchored in the apical membrane of the epithelial cells of the thick ascending limb of the loop of Henle, where it undergoes proteolytic cleavage to be released in urine.6,15 Disease manifestations related to mutant UMOD are secondary to toxic gain of function, with endoplasmic reticulum accumulation and reduced urinary levels as the primary molecular defect.2,6,15–18 So far, only two deletions in the UMOD gene, respectively involving 27 and 99 nucleotide pairs, and both affecting evolutionarily conserved residues in exon 4, have been associated with ADTKD/FJHN/MCKD.5,19 Although the size of the UMOD deletion identified in our family is much larger than the two formerly described, it does not disrupt any of the structural domains or remove critical amino acid residues of uromodulin.6,15,20 Since UMOD exon 11 encodes for the last amino acids of the C-terminal of the precursor protein, downstream from the anchoring site, as well as for the 3′-untranslated region (3′UTR), containing the polyadenylation signal sequence and the polyadenylation site, we postulate that its deletion might be pathogenic through loss of critical regulatory 3′UTR mRNA functions,21 representing a novel pathogenic mechanism for ADTKD-UMOD.
The SLC8A1 gene encodes the sodium/calcium exchanger 1 (NCX1), an ubiquitously expressed protein with a diversity of tissue-specific splice variants, belonging to the calcium/cation antiporter superfamily.8 Altered expression and regulation of NCX1 proteins contribute to disturbed calcium homeostasis in cardiovascular disease (including hypertension, heart failure, arrhythmia and cerebral ischemia), diabetes, immune response, and tumor angiogenesis, as well as to impaired calcium reabsorption in the kidney.8 In our family, heterozygosity for the SLC8A1 p.(Arg6*) allele was consistently associated with earlier clinical manifestations of ADTKD and progression to kidney failure (KF) requiring KRT. Since the natural history of ADTKD is more severe in males,22 the fact that all three double heterozygous patients were males and the patient who inherited only the UMOD exon 11 deletion was a female needs consideration when interpreting those correlations. Unfortunately, on family screening, we could not identify any subject heterozygous only for the SLC8A1 p.(Arg6*) allele, or a double heterozygous female for both the SLC8A1 p.(Arg6*) allele and the UMOD exon 11 deletion, which would have helped establishing more robust genotype–phenotype correlations. However, the age difference at the initiation of KRT between the proband's father and aunt was much larger than expected from gender-specific survival curves without KF for ADTKD-UMOD,22 an observation suggesting that the SLC8A1 p.(Arg6*) allele exerted a pathogenic role in this family, at least as a disease modifier.
In conclusion: genotyping analysis of a Portuguese family diagnosed with FJHN/ADTKD led to identifying a novel, large, terminal single-exon deletion in UMOD, cosegregating with an ultra-rare nonsense SLC8A1 variant in most of the affected individuals, with the double heterozygous patients exhibiting a more severe clinical phenotype. These findings require further investigation to elucidate the molecular mechanism(s) underlying the pathogenicity of UMOD exon 11 deletion, which does not encode any of the critical structural domains or amino acid residues or uromodulin; as well as to clarify the role of the SLC8A1 locus in ADTKD.
FundingThis research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of interestThe authors have no conflict of interest to disclose.
Results of this family study were presented as a poster (#5302) at the 60th Congress of the European Renal Association, that took place on June 15-18, 2023, in Milan (Italy), and published in the Abstract Book — Nephrol Dial Transplant. 2023;38 (Suppl. 2):i236-237.



