



The UMOD gene encodes the uromodulin protein, which plays a crucial role in renal function. Genetic alterations affecting its correct function are mainly related to Autosomal Dominant Tubulointerstitial Kidney Disease (ADTKD), progressive renal failure and hyperuricaemia, among other variable clinical phenotypes. In the Galician population there are recurrent mutations in this gene, this study aims to phenotypically characterize the recurrent variants to improve the prognosis and management strategies of affected patients.
MethodsIn a Galician population characterized by high genetic conservation, a retrospective cohort study was conducted with 37 patients from 15 families carrying recurrent variants in UMOD (p.C255Y and p.Q316P, from transcript NM_001008389.3). Clinical data were collected, including renal function, hyperuricemia, hypertension and presence of renal cysts. Genomic analyses were performed by NGS and Sanger sequencing, variant classification were conducted according to ACMG guidelines. Statistical comparisons were performed using Mann-Whitney, Chi-square and Fisher tests, with Benjamini-Hochberg correction for multiple testing.
ResultsThe cohort included 28 carriers of p.C255Y and 9 of p.Q316P genetic variants. Both variants affect highly conserved domains with low tolerance to amino acid changes, which alters protein function and has clinical effects in patients. Hyperuricemia was observed in 76% of p.C255Y carriers and 50% of p.Q316P carriers, while interestingly only the first variant was associated with episodes of gout. Renal cysts and hypertension were identified in about half of the patients, independently of variant type. Kaplan–Meier curves suggested an earlier progression to hyperuricemia and advanced chronic kidney disease (CKD) in p.C255Y carriers, although without reaching statistical significance.
ConclusionsRecurrent UMOD mutations in a Galician cohort revealed shared clinical features, including hyperuricemia and CKD progression, with phenotypic variability influenced by age and additional genetic modifiers. The findings highlight the prognostic value of genotype-phenotype correlations and the need for tailored clinical management in ADTKD patients.
El gen UMOD codifica la proteína uromodulina, la cual tiene una función crucial en el funcionamiento renal. Las alteraciones genéticas que afectan a su correcta función se relacionan principalmente con nefropatía tubulointersticial autosómica dominante (ADTKD), insuficiencia renal progresiva e hiperuricemia, entre otros fenotipos clínicos variables. En la población gallega existen mutaciones recurrentes en este gen, este estudio tiene como objetivo caracterizar fenotípicamente las variantes recurrentes para mejorar el pronóstico y las estrategias de manejo de los pacientes afectados.
MetodologíaSe realizó un estudio de cohorte retrospectivo con 37 pacientes de 15 familias portadoras de variantes recurrentes en UMOD (p.C255Y y p.Q316P, del transcrito NM_001008389.3) en una población gallega caracterizada por su alta conservación genética. Se recopilaron datos clínicos, incluyendo función renal, hiperuricemia, hipertensión y presencia de quistes renales. Los análisis genómicos se realizaron mediante secuenciación masiva (NGS) y Sanger, clasificando las variantes según las guías del ACMG. Las comparaciones estadísticas se llevaron a cabo mediante pruebas de Mann-Whitney, Chi-cuadrado y Fisher, con corrección de Benjamini-Hochberg para pruebas múltiples.
ResultadosLa cohorte incluyó 28 portadores de p.C255Y y 9 de p.Q316P. Ambas variantes afectan dominios altamente conservados con baja tolerancia a los cambios de aminoácidos, lo que altera la función de la proteína y tiene efectos clínicos en los pacientes. Se observó hiperuricemia en el 76% de los portadores de p.C255Y y en el 50% de los portadores de p.Q316P, mientras que sólo se asoció la primera variante con episodios de gota. Los quistes renales y la hipertensión fueron identificados en aproximadamente la mitad de los pacientes, independientemente del tipo de variante. Las curvas de Kaplan–Meier sugirieron una progresión más temprana a hiperuricemia y a enfermedad renal crónica avanzada (ERCA) en portadores de p.C255Y, aunque sin alcanzar significancia estadística.
ConclusionesLas mutaciones recurrentes de UMOD en una cohorte gallega revelaron características clínicas compartidas, como hiperuricemia y progresión a ERCA, con una variabilidad fenotípica influenciada por la edad y modificadores genéticos adicionales. Estos hallazgos subrayan el valor pronóstico de las correlaciones genotipo-fenotipo y la necesidad de un manejo clínico individualizado en pacientes con ADTKD.
Chronic kidney disease (CKD) is a serious disease associated with high morbidity and mortality and increasing health expenditure per patient. However, CKD etiology is often not firmly established and, in many patients, remains unknown.1 The etiology of CKD can be due to either environmental, genetic factors and/or a combination of both.2,3 Renal diseases due to genetic factors may follow an autosomal dominant or recessive pattern of inheritance.
For years a group of autosomal dominant renal diseases has been described that mainly affects tubular and cystic level, characterized by progressive deterioration of renal function accompanied by tubulointerstitial fibrosis, being UMOD one of the genes responsible for these diseases.
UMOD presentation, function and protein structureThis work is focused on the phenotypic manifestations caused by alterations in UMOD gene. This gene encodes the uromodulin protein (also known as Tamm-Horsfall glycoprotein), and is specifically expressed at renal level.4
Under physiological conditions, uromodulin is the most abundant protein in urine, and it is secreted by the epithelial cells of the thick ascending limb of loop of Henle in a bidirectional manner into the urine and circulation.4 Uromodulin can be found polymerized forming filaments, or in its non-polymeric form.4 This non-polymeric form has different renal and systemic functions, this is why its site of action is important 4,5 The main functions of this protein are regulation of ionic transport in the kidney, urinary and systemic homeostasis, as highlighted by Trachtman et al.6 in their study. In addition, uromodulin has a protective effect against calcium nephrolithiasis and urinary tract infections, with a crucial role in local and systemic immunomodulation.7–9
Its protein structure differentiates 4 EGF-like domains (EGF-like I, EGF-like II, EGF-like III and EGF-like IV), a domain with 10 conserved cystines (D10C), and a bipartite zona pellucida (ZP) in ZP-N and ZP-C.10–14 These domains allow junctions that make the protein to fold three-dimensionally and form its functional structure and polymerization.7,11,12,14,15
Molecular impact of genetic alterations in UMODMutations in UMOD gene can cause defects in the structure, maturation, secretion and/or function of uromodulin, depending on the type of variant and affected domain . Most of pathogenic alterations in this gene affect the correct structural conformation, resulting in an abnormal protein. This anomalous protein is retained and accumulated in the endoplasmic reticulum, causing cell death in the renal tubule.7,16,17 This phenomenon can trigger endoplasmic reticulum stress, activation of inflammatory responses, renal interstitial fibrosis and secondary mitochondrial dysfunction,7,18 together with compensatory reduced expression of the sodium-potassium-chloride cotransporter (Na+–K+–2Cl–) (NKCC2),7,13,19 contributes to increased proximal urate reabsorption and progressive deterioration of renal function due to the oxidative stress associated with the accumulation of this abnormal protein, leading to hyperuricemia, episodes of gout, fibrosis and renal failure7,18 (Fig. 1).
.7 NKCC2: sodium–potassium–chlorine cotransporter (Na+–K+–2Cl–); ER: endoplasmic reticulum; uUMOD: UMOD in urine; UPR: unfolded protein response.")
Schematic diagram of the pathophysiology of UMOD alterations. On the left, the restricted expression of UMOD to the ascending branch of Henle and the first sections of the distal convoluted tubule is depicted. On the right, the altered processes due to expression of mutant UMOD. Source: adapted from Mabillard et al. (2023).7 NKCC2: sodium–potassium–chlorine cotransporter (Na+–K+–2Cl–); ER: endoplasmic reticulum; uUMOD: UMOD in urine; UPR: unfolded protein response.
In the literature, a great phenotypic variability has been associated with alterations in UMOD, even among individuals who are carriers of the same alteration. However, there are other genetic and environmental modifying factors that may influence the presentation and progression of the disease.
Patients with alterations in UMOD, may present a decrease in the fractional excretion of urate, which causes hyperuricemia, and sporadic glomerular involvement with manifestations such as microhematuria. Proteinuria is usually mild or absent. The presence of renal cysts has also been observed in patients with alterations in this gene, although to a much lesser extent than other manifestations such as hiperuricemia.20–24
It has been observed that individuals with mutations in UMOD reach end-stage renal disease between the ages of 25 and 70 years or more, and according to the study by Ayasreh et al.25,26, those with a history of gout experienced its onset between the ages of 3 and 51 years.
Clinical studies have shown that kidney biopsies from affected patients reveal interstitial fibrosis and tubular atrophy, confirming the pattern of tubulointerstitial damage associated with these mutations.27
ADTKD-UMOD. Main hereditary kidney disease associated with UMODGenes associated with Autosomal Dominant Tubulointerstitial Disease (ADTKD) development are HNF1B, MUC1, REN, SEC61A1, and UMOD. All of them exhibit phenotypic variability (diabetes, liver cancer, renal cysts, and psychiatric disorders, among others), with ADTKD being the common manifestation that links them, but with incomplete penetrance. Variants identified in the UMOD gene are mostly associated with ADTKD.5,7,28–31 Patients with ADTKD-UMOD show a progressive course of kidney failure with early onset of hyperuricemia, and although the vast majority of cases are asymptomatic, the development of gout has also been described.28,30–32
Recurrent UMOD mutationsRecent studies in closed populations have identified several recurrent mutations in UMOD. These recurrent mutations are of particular interest due to their high prevalence in specific populations and their association with severe clinical phenotypes.7 The study of recurrent mutations offers a unique window to understand the pathogenic mechanisms underlying the function of the altered gene, characterize the phenotypic manifestations, establishing a correlation between the genetic mutation and disease progression, and develop targeted therapeutic strategies.
An association has been described between the age of onset of end-stage renal disease, the underlying UMOD mutation and the domain in which the mutation is found.32 In this sense, analyzing mutations in UMOD and their relationship to inherited kidney disease provides a solid basis for early identification, genetic diagnosis and clinical management of these patients.
In the last 25 years, our team has focused on studying and establishing the genetic map of hereditary diseases in Galicia divided into 3 large groups: cystic disease, glomerular disease and tubulointerstitial disease. This comprehensive study includes 2538 families and 4094 individuals studied genetically, with the aim of establishing a genotype-phenotype correlation. Within the group of tubulointerstitial diseases, 371 families were genetically characterized and 81 of them with ADTKD, of which 31 are families with mutations in UMOD. This study identifies the presence in the Galician population of 2 recurrent genetic variants responsible for most cases of ADTKD without familial relationship (UMOD, transcript NM_001008389.3: p.C255Y and p.Q316P, transcribed NM_001008389.3). In this article a genotype-phenotype correlation is established in 37 patients from 15 unrelated families in the Galician community, in order to establish phenotypic patterns associated with the presence of these variants.
MethodologyStudy participants and clinical characterizationThis is a retrospective, multicenter, observational cohort study that analyzed NefroCHUS cohort of 4094 patients with suspected hereditary kidney diseases. This cohort includes patients from different hospitals throughout Spain, with an enrichment in the Galician population.
Inclusion criteria for patients' selection: 1) genetic testing requested by a nephrologist; 2) initial clinical suspicion of ADTKD; 3) genetic sequencing of genes associated with ADTKD (HNF1B, MUC1, REN, SEC61A1, and UMOD) at least; and 4) patients carrying one of the two most recurrent mutations of UMOD in Galician community: p.C255Y or p.Q316P.
A total of 37 patients from 15 families were selected. Ten of these families carried p.C255Y mutation, and the remaining five carried p.Q316P.
For the clinical characterization of the patients in this study, clinical data such as hypertension, estimated glomerular filtration rate (eGFR), CKD stage, hyperuricemia (considered when serum uric acid levels were greater than 7 mg/dl), fractional excretion of uric acid, gout episode, presence of renal cysts, albuminuria, hematuria, type of renal replacement therapy and kidney transplant were collected.
Molecular analysisPatient DNA was extracted from peripheral blood leukocytes using the commercial Chemagic Blood 1k-3k kits using the Chemagen extraction robot (PerkinElmer, Waltham, Massachusetts, USA) and the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany), following the manufacturers' recommended protocols. Quality and concentration of the extracted DNA was assessed using the Qubit 3 fluorometer, Broad Range kit (Thermo Fisher Scientific, Waltham, Massachusetts, USA).
All patients included in this selection underwent genetic testing, either using Next-Generation Sequencing (NGS) or targeted sequencing using Sanger sequencing. Whenever technology allowed, the index cases in each family were genetically tested using NGS technology (in 86.67% of index cases, 13/15 probands).
The NGS technique used in the patients in this study is the sequencing of a panel of known genes associated with the most prevalent tubular and cystic renal pathologies33. Obtaining the sequence of the exons and flanking regions of selected genes. This identifies point variants, small insertions/deletions (indels), and the presence of structural variants (copy number variants [CNVs]). Raw sequencing reads were processed according to GATK best practice guidelines and aligned to the human reference genome (GRCh37) using bwa version 0.7.17-r1188. Low-quality reads were removed from the primary dataset using fastp version 0.20. Variant calling was performed using GATK version 4.1.9, Pindel version 0.2.5b9, and ExomeDepth version 1.1.15, in accordance with best practice recommendations. Variant annotation was performed using an in-house process, combining functional gene annotations from SnpEff and ANNOVAR to retrieve population frequencies (1000 Genomes Project, gnomAD, and an in-house database, among others), functional prediction scores (SIFT, CADD, etc.), and clinical information from databases such as ClinVar and OMIM.
Sanger sequencing is a targeted sequencing technique that sequences short DNA regions of interest with high accuracy. This technique is used in most relatives of index cases to study the candidate alteration segregation. By identifying the candidate variant that could cause pathology in index cases, sequencing can be targeted to that region and more quickly identify whether relatives are carriers of the variant.
Characterization of identified mutationsThe variants were classified independently by two geneticists specialized in hereditary kidney diseases following the guidelines and recommendations of the American College of Medical Genetics (ACMG).34
A literature search was conducted for the 2 recurrent variants in this study using different search tools such as PubMed, ClinVar, Human Gene Mutation Database (HGMD) and MetaDome to determine whether they have been previously reported, their molecular involvement and their phenotypic associations.
Statistical analysisThe statistical significance of the results was assessed for nominal variables using the Chi-square test or Fisher's exact test for variables with a frequency greater or less than 5, respectively, and for quantitative variables, the Mann-Whitney-Wilcoxon test using RStudio. Due to the small sample size, p-values were adjusted using the Benjamini-Hochberg method to control the false discovery rate (FDR). This adjusted p-value is the q-value; statistical significance is considered a q-value greater than 0.05.
The log-rank statistical test was applied to the Kaplan–Meier survival curves to study the differences between variant types and establish the p-value.
ResultsThis study selected and analyzed 37 patients from 15 families with Autosomal Dominant Renal Tubulointerstitial Disease (ADTKD) carrying mutations in the UMOD gene from different hospitals in Galicia. These patients were divided into two subcohorts. Of these, 10 probands and 18 relatives had the p.C255Y mutation (all individuals carry the alteration in heterozygosity, except for one relative (F4.S3) who carries the variant in homozygosity), while 5 probands and 4 relatives carry the p.Q316P mutation (in heterozygosity). The age distributions in both groups are similar (Fig. 2A), making the clinical manifestations of both subcohorts comparable.
 Age distribution in the UMOD recurrent variant cohort divided by variant type. The medians of age in both groups are very similar, and no statistically significant differences are seen between the 2 groups (ns), with a p-value of 0.9222. B) Representation of the diagnosis of hyperuricemia as a function of age for each variant using a Kaplan–Meier survival curve, with the log-rank test calculating a p-value of 0.1040, so that the differences are not statistically significant. C) Representation of patients in ACKD according to age for each variant by means of a Kaplan–Meier survival curve, with the log-rank test calculating a p-value of 0.4156, so the differences are not statistically significant. D) Patients in each stage of CKD divided by type of alteration.")
A) Age distribution in the UMOD recurrent variant cohort divided by variant type. The medians of age in both groups are very similar, and no statistically significant differences are seen between the 2 groups (ns), with a p-value of 0.9222. B) Representation of the diagnosis of hyperuricemia as a function of age for each variant using a Kaplan–Meier survival curve, with the log-rank test calculating a p-value of 0.1040, so that the differences are not statistically significant. C) Representation of patients in ACKD according to age for each variant by means of a Kaplan–Meier survival curve, with the log-rank test calculating a p-value of 0.4156, so the differences are not statistically significant. D) Patients in each stage of CKD divided by type of alteration.
The appearance of hyperuricemia is one of the most representative characteristics of the clinical diagnosis of this disease; therefore, the relationship between hyperuricemia and onset age was studied for each of the variants using a Kaplan–Meier survival curve (Fig. 2B). Statistical analysis of the differences between the two variables using the log-rank test, resulted in a p-value of 0.1040.
The progression to CKD is another characteristic of UMOD patients, due to their progressive deterioration of kidney function. This is why Fig. 2C shows that CKD does not occur in any case under the age of 38, and from this age onwards, the survival curve for both abnormalities declines.
Fig. 2D represents the distribution of the different stages of CKD in each subcohort. Although no statistically significant differences were observed in the CKD stage (Table 1), Fig. 2D shows that, while patients carrying the p.C255Y variant are more frequently present in stage 3b, the p.Q316P carriers are less frequently present in this stage. Therefore, there could be a trend with respect to disease progression.
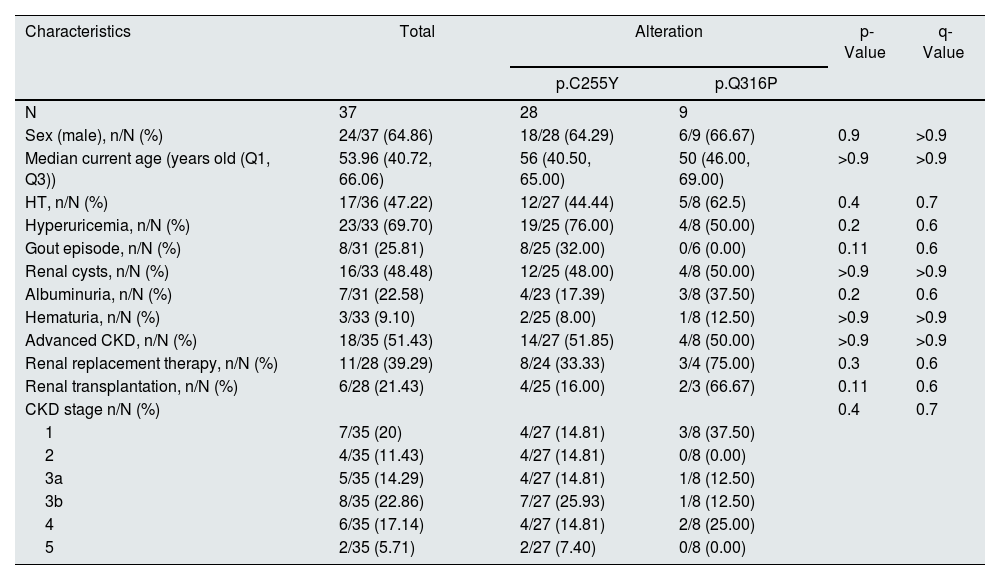
Statistical description of the clinical manifestations of patients with recurrent UMOD alterations according to their alteration.
| Characteristics | Total | Alteration | p-Value | q-Value | |
|---|---|---|---|---|---|
| p.C255Y | p.Q316P | ||||
| N | 37 | 28 | 9 | ||
| Sex (male), n/N (%) | 24/37 (64.86) | 18/28 (64.29) | 6/9 (66.67) | 0.9 | >0.9 |
| Median current age (years old (Q1, Q3)) | 53.96 (40.72, 66.06) | 56 (40.50, 65.00) | 50 (46.00, 69.00) | >0.9 | >0.9 |
| HT, n/N (%) | 17/36 (47.22) | 12/27 (44.44) | 5/8 (62.5) | 0.4 | 0.7 |
| Hyperuricemia, n/N (%) | 23/33 (69.70) | 19/25 (76.00) | 4/8 (50.00) | 0.2 | 0.6 |
| Gout episode, n/N (%) | 8/31 (25.81) | 8/25 (32.00) | 0/6 (0.00) | 0.11 | 0.6 |
| Renal cysts, n/N (%) | 16/33 (48.48) | 12/25 (48.00) | 4/8 (50.00) | >0.9 | >0.9 |
| Albuminuria, n/N (%) | 7/31 (22.58) | 4/23 (17.39) | 3/8 (37.50) | 0.2 | 0.6 |
| Hematuria, n/N (%) | 3/33 (9.10) | 2/25 (8.00) | 1/8 (12.50) | >0.9 | >0.9 |
| Advanced CKD, n/N (%) | 18/35 (51.43) | 14/27 (51.85) | 4/8 (50.00) | >0.9 | >0.9 |
| Renal replacement therapy, n/N (%) | 11/28 (39.29) | 8/24 (33.33) | 3/4 (75.00) | 0.3 | 0.6 |
| Renal transplantation, n/N (%) | 6/28 (21.43) | 4/25 (16.00) | 2/3 (66.67) | 0.11 | 0.6 |
| CKD stage n/N (%) | 0.4 | 0.7 | |||
| 1 | 7/35 (20) | 4/27 (14.81) | 3/8 (37.50) | ||
| 2 | 4/35 (11.43) | 4/27 (14.81) | 0/8 (0.00) | ||
| 3a | 5/35 (14.29) | 4/27 (14.81) | 1/8 (12.50) | ||
| 3b | 8/35 (22.86) | 7/27 (25.93) | 1/8 (12.50) | ||
| 4 | 6/35 (17.14) | 4/27 (14.81) | 2/8 (25.00) | ||
| 5 | 2/35 (5.71) | 2/27 (7.40) | 0/8 (0.00) | ||
P-values compare the 2 recurrent alterations using the Mann-Whitney-Wilcoxon test for quantitative data and the Chi-square test or Fisher exact test for variables with a frequency of more or less than 5, respectively, for categorical data; q-values are the p values corrected with the Benjamini-Hochberg test (correction of the false discovery rate for multiple testing). CKD: Chronic Kidney Disease; HT: hypertension; n: represents the number of subjects with the characteristic; N: represents the number of total subjects with information on that characteristic.
Table 1 represents the descriptive statistics of the characteristics observed in the patients of this cohort along with their proportions. Table 2 details the characteristics observed in each patient.
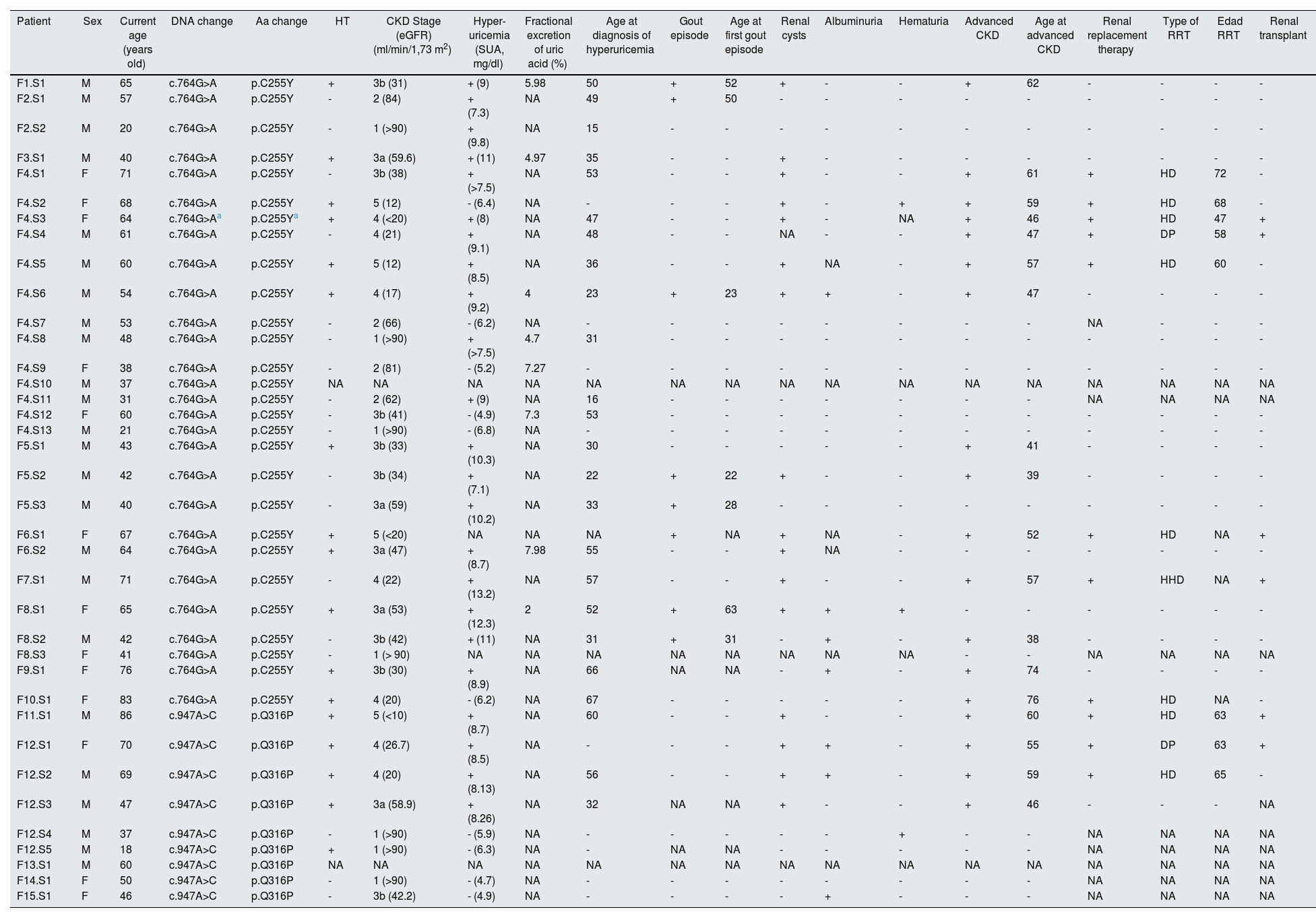
Phenotypic characteristics of the cohort of patients with recurrent UMOD alterations.
| Patient | Sex | Current age (years old) | DNA change | Aa change | HT | CKD Stage (eGFR) (ml/min/1,73 m2) | Hyper-uricemia (SUA, mg/dl) | Fractional excretion of uric acid (%) | Age at diagnosis of hyperuricemia | Gout episode | Age at first gout episode | Renal cysts | Albuminuria | Hematuria | Advanced CKD | Age at advanced CKD | Renal replacement therapy | Type of RRT | Edad RRT | Renal transplant |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1.S1 | M | 65 | c.764G>A | p.C255Y | + | 3b (31) | + (9) | 5.98 | 50 | + | 52 | + | - | - | + | 62 | - | - | - | - |
| F2.S1 | M | 57 | c.764G>A | p.C255Y | - | 2 (84) | + (7.3) | NA | 49 | + | 50 | - | - | - | - | - | - | - | - | - |
| F2.S2 | M | 20 | c.764G>A | p.C255Y | - | 1 (>90) | + (9.8) | NA | 15 | - | - | - | - | - | - | - | - | - | - | - |
| F3.S1 | M | 40 | c.764G>A | p.C255Y | + | 3a (59.6) | + (11) | 4.97 | 35 | - | - | + | - | - | - | - | - | - | - | - |
| F4.S1 | F | 71 | c.764G>A | p.C255Y | - | 3b (38) | + (>7.5) | NA | 53 | - | - | + | - | - | + | 61 | + | HD | 72 | - |
| F4.S2 | F | 68 | c.764G>A | p.C255Y | + | 5 (12) | - (6.4) | NA | - | - | - | + | - | + | + | 59 | + | HD | 68 | - |
| F4.S3 | F | 64 | c.764G>Aa | p.C255Ya | + | 4 (<20) | + (8) | NA | 47 | - | - | + | - | NA | + | 46 | + | HD | 47 | + |
| F4.S4 | M | 61 | c.764G>A | p.C255Y | - | 4 (21) | + (9.1) | NA | 48 | - | - | NA | - | - | + | 47 | + | DP | 58 | + |
| F4.S5 | M | 60 | c.764G>A | p.C255Y | + | 5 (12) | + (8.5) | NA | 36 | - | - | + | NA | - | + | 57 | + | HD | 60 | - |
| F4.S6 | M | 54 | c.764G>A | p.C255Y | + | 4 (17) | + (9.2) | 4 | 23 | + | 23 | + | + | - | + | 47 | - | - | - | - |
| F4.S7 | M | 53 | c.764G>A | p.C255Y | - | 2 (66) | - (6.2) | NA | - | - | - | - | - | - | - | - | NA | - | - | - |
| F4.S8 | M | 48 | c.764G>A | p.C255Y | - | 1 (>90) | + (>7.5) | 4.7 | 31 | - | - | - | - | - | - | - | - | - | - | - |
| F4.S9 | F | 38 | c.764G>A | p.C255Y | - | 2 (81) | - (5.2) | 7.27 | - | - | - | - | - | - | - | - | - | - | - | - |
| F4.S10 | M | 37 | c.764G>A | p.C255Y | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| F4.S11 | M | 31 | c.764G>A | p.C255Y | - | 2 (62) | + (9) | NA | 16 | - | - | - | - | - | - | - | NA | NA | NA | NA |
| F4.S12 | F | 60 | c.764G>A | p.C255Y | - | 3b (41) | - (4.9) | 7.3 | 53 | - | - | - | - | - | - | - | - | - | - | - |
| F4.S13 | M | 21 | c.764G>A | p.C255Y | - | 1 (>90) | - (6.8) | NA | - | - | - | - | - | - | - | - | - | - | - | - |
| F5.S1 | M | 43 | c.764G>A | p.C255Y | + | 3b (33) | + (10.3) | NA | 30 | - | - | - | - | - | + | 41 | - | - | - | - |
| F5.S2 | M | 42 | c.764G>A | p.C255Y | - | 3b (34) | + (7.1) | NA | 22 | + | 22 | + | - | - | + | 39 | - | - | - | - |
| F5.S3 | M | 40 | c.764G>A | p.C255Y | - | 3a (59) | + (10.2) | NA | 33 | + | 28 | - | - | - | - | - | - | - | - | - |
| F6.S1 | F | 67 | c.764G>A | p.C255Y | + | 5 (<20) | NA | NA | NA | + | NA | + | NA | - | + | 52 | + | HD | NA | + |
| F6.S2 | M | 64 | c.764G>A | p.C255Y | + | 3a (47) | + (8.7) | 7.98 | 55 | - | - | + | NA | - | - | - | - | - | - | - |
| F7.S1 | M | 71 | c.764G>A | p.C255Y | - | 4 (22) | + (13.2) | NA | 57 | - | - | + | - | - | + | 57 | + | HHD | NA | + |
| F8.S1 | F | 65 | c.764G>A | p.C255Y | + | 3a (53) | + (12.3) | 2 | 52 | + | 63 | + | + | + | - | - | - | - | - | - |
| F8.S2 | M | 42 | c.764G>A | p.C255Y | - | 3b (42) | + (11) | NA | 31 | + | 31 | - | + | - | + | 38 | - | - | - | - |
| F8.S3 | F | 41 | c.764G>A | p.C255Y | - | 1 (> 90) | NA | NA | NA | NA | NA | NA | NA | NA | - | - | NA | NA | NA | NA |
| F9.S1 | F | 76 | c.764G>A | p.C255Y | + | 3b (30) | + (8.9) | NA | 66 | NA | NA | - | + | - | + | 74 | - | - | - | - |
| F10.S1 | F | 83 | c.764G>A | p.C255Y | + | 4 (20) | - (6.2) | NA | 67 | - | - | - | - | - | + | 76 | + | HD | NA | - |
| F11.S1 | M | 86 | c.947A>C | p.Q316P | + | 5 (<10) | + (8.7) | NA | 60 | - | - | + | - | - | + | 60 | + | HD | 63 | + |
| F12.S1 | F | 70 | c.947A>C | p.Q316P | + | 4 (26.7) | + (8.5) | NA | - | - | - | + | + | - | + | 55 | + | DP | 63 | + |
| F12.S2 | M | 69 | c.947A>C | p.Q316P | + | 4 (20) | + (8.13) | NA | 56 | - | - | + | + | - | + | 59 | + | HD | 65 | - |
| F12.S3 | M | 47 | c.947A>C | p.Q316P | + | 3a (58.9) | + (8.26) | NA | 32 | NA | NA | + | - | - | + | 46 | - | - | - | NA |
| F12.S4 | M | 37 | c.947A>C | p.Q316P | - | 1 (>90) | - (5.9) | NA | - | - | - | - | - | + | - | - | NA | NA | NA | NA |
| F12.S5 | M | 18 | c.947A>C | p.Q316P | + | 1 (>90) | - (6.3) | NA | - | NA | NA | - | - | - | - | - | NA | NA | NA | NA |
| F13.S1 | M | 60 | c.947A>C | p.Q316P | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| F14.S1 | F | 50 | c.947A>C | p.Q316P | - | 1 (>90) | - (4.7) | NA | - | - | - | - | - | - | - | - | NA | NA | NA | NA |
| F15.S1 | F | 46 | c.947A>C | p.Q316P | - | 3b (42.2) | - (4.9) | NA | - | - | - | - | + | - | - | - | NA | NA | NA | NA |
Aa, amino acid; CKD, chronic kidney disease; HD, hemodialysis; HHD, home hemodialysis; HT, hypertension; eGFR, estimated glomerular filtration rate (calculated with CKD-EPI); F, female; NA, no data available; SUA, serum uric acid; RRT, renal replacement therapy; M, male; PD, peritoneal dialysis.
At molecular level, the p.C255Y alteration is classified as probably pathogenic. It affects the D10C10 domain; this domain is highly conserved, so genetic alterations affecting this domain are highly likely to impact its function.13–15 When studying tolerance to the change of reference amino acid at position 255 using MetaDome program,35 we found that it is intolerant to change, with a tolerance score of 0.22 (scores lower than 0.7 are considered intolerant to amino acid changes). Therefore, this recurrent alteration, which changes the cystine residue to that a tyrosine, has a negative impact at protein level.35
This is the most recurrent UMOD variant in our Galician cohort of patients with suspected hereditary kidney disease, with a total of 28 carriers. Of these, 64.29% are men (18/28).
Twelve patients (44.44%) presented with hypertension (HT), either at the time of diagnosis or during the course of the disease. Furthermore, 77.77% of patients had some degree of CKD (considered to be stage 2 or higher) at the time of the study, with a mean age of 57 years. Eight patients (33.33%) progressed to End-Stage Renal Disease (ESRD), starting dialysis at a mean age of 61 years.
Of note is the case of the female patient F4.S3, a homozygous carrier of the p.C255Y alteration. F4.S3 began renal replacement therapy (RRT) at 46 years old, a decade or more earlier than the rest of the patients with RRT in the cohort (Table 2). This suggests that alterations in homozygosity are associated with more accelerated and severe progression of kidney disease.
Seventy-six percent of cases met the criteria of hyperuricemia (19 of 25 patients). Hyperuricemia diagnosis and initiation of treatment with allopurinol occurred at a mean age of 41.38 years (range: 16–67 years). Urinary sediment was generally unremarkable, with only 4 cases of albuminuria (17.39%) and 2 cases of microhematuria (8%). Furthermore, cyst development was observed in 48% of patients.
Among the 32% of patients with gout, the mean age at onset of the first gout episode was 38.43 years (range: 22–63 years). Fig. 3 shows the survival curve of the ages at the first gout episode. Despite the onset in early adulthood, several elderly patients have not developed gout, which could be interpreted as indicating that this event is not highly related to age.
Intrafamilial variability of p.C255Y. Family F4
Of these carrier patients, 13 belong to family F4, which has a history of hyperuricemia and ESRD, conditions likely associated with this mutation. Two individuals in this family have undergone a kidney transplant. At intrafamilial level, we observed characteristics with a high familial prevalence, such as an estimated glomerular filtration rate lower than 90 ml/min/1.73 m2, indicative of kidney disease.
Additionally, it has been found variability in the development of hyperuricemia, with this characteristic being observed in patients of different ages.
It was also observed some age-related variability in other characteristics, with approximately half of the older relatives presenting the trait whereas the other half of younger relatives do not. These characteristics include: the onset of advanced CKD (individuals under 54 years of age in this family have not progressed), RRT (the cut-off age for this trait is 60 years), and presence of renal cysts (with a cutting age of 54 years).
It is also worth noting that six members of this family are also carriers of a heterozygous pathogenic alteration in PKHD1gene, c.383delC, p.Thr128fs (F4.S1, F4.S2, F4.S6, F4.S7, F4.S9, and F4.S13). Alterations in this gene are primarily associated with the development of renal cysts, with a primarily autosomal recessive inheritance, although a cystic phenotype has also been described in patients with heterozygous alterations.36 In the family F4, there are a total of 5 individuals with renal cysts, 3 of whom are carriers of the PKHD1 alteration. Therefore, we cannot confirm or rule out that this alteration in PKHD1 modulates the appearance of renal cysts in patients who are carriers of alterations in UMOD and PKHD1.
UMOD c.947A>C alteration (p.Q316P)UMOD p.Q316P alteration is also molecularly classified as likely pathogenic. It affects the EGF-like domain IV.10–12 This domain is highly conserved and it is involved in the bisulfate bonds that contribute to the three-dimensional structure of uromodulin.10 This position is also intolerant to change, with a tolerance score of 0.3637.
This variant was found in 9 patients, of whom 66.67% were men (6/9). In this cohort, it was observed a higher percentage of patients with hypertension, present in 5 patients (62.5%). At the time of diagnosis, 50% of patients had some degree of CKD (considered from stage 2 onward), at a mean age of 54 years. To date, 3 patients (75%) have achieved ESRD, with onset on RRT at a mean age of 63.67 years.
Fifty percent of the cohort met criteria for hyperuricemia. Diagnosis of hyperuricemia and initiation of allopurinol treatment occurred at a mean age of 49.33 years (range: 32–60 years). To date, no episodes of gout have been reported in these patients, although there is a family history of gout.
The urinary sediment of this second cohort was also unremarkable, with microalbuminuria present in 3 out of the 8 patients (one of them with albumin creatinine ratio> 300 mg/g) and only one case of microhematuria. Similar to the first subcohort, cystic development was observed in 50% of patients.
Intrafamilial variability of p.Q316P. Family F12Family F12 has the highest number of p.Q316P carriers, includes 5 patients, and 4 of these present hypertension. The effect of age on renal impairment is also evident in this family, as the two youngest members of the family only present a pathological phenotype of hypertension, without any additional characteristics, while the older patients have a glomerular filtration rate of 35.2 ml/min/1.73 m2; renal cysts; hyperuricemia; and have reached advanced CKD.
DiscussionThis is a retrospective study of recurrent variants in UMOD in a genetically closed Galician population cohort. The study of the Galician population is of high value in terms of the characterization of recurrent variants and genotype-phenotype correlations, since the region of Galicia is geographically located in western Spain, and geographic barriers have provided a high level of genetic conservation, ethnicity and consanguinity.37,38
The alterations studied are located in positions intolerant to change,35 highly conserved10–12 and with a structural function,10,13–15 which is why, although it is an alteration that only changes one amino acid for another, the variants have a pathogenic effect, and result in renal manifestations in patients, since the protein cannot be correctly structured three-dimensionally, and its function is altered.
With the clinical data obtained from this cohort, it is notable that these alterations in UMOD have certain common phenotypes, confirming the important role of the gene in processes such as uric acid transport and absorption, cyst formation and renal function, which can lead over the years to early renal impairment.
Of the entire cohort, only F4.S3 carries the p.C255Y alteration in homozygosis, both in this study and in other previous studies it has been observed that biallelic alterations in this gene produce a more aggressive and earlier development of the clinical manifestations.39–43
Despite the difference in size of the 2 subcohorts, certain similarities and differences can be appreciated between the patients carrying them. Among the similarities, and in line with the literature on the phenotype associated with the alterations in UMOD, more than half of the patients present hyperuricemia (76 and 50%, respectively). The presence of renal cysts and hypertension was observed in approximately half of the carriers for both disorders. Approximately half of the patients developed advanced CKD (with greater representation in the older population) and required renal replacement therapy.
The study by Moskowitz et al.32 compares the phenotype of various alterations in UMOD, including p.C255Y and p.Q316P. These patients mainly present hyperuricemia, gout (in a smaller proportion) and reach ESRD. In that study it was observed that the alteration with which the patients reached ESRD later was p.Q316P.32 In our cohort, no statistical differences between the alterations were observed. Even so (Fig. 2C), carriers of p.Q316P took longer to reach advanced CKD, the first case being at 46 years of age, whereas the first case of carriers of p.C255Y was at 38 years. It should be noted that this characteristic is related to renal deterioration due to multiple factors and is directly associated with age.
Fig. 2B shows that the presence of hyperuricemia is a characteristic that can occur at different stages of life; in some patients it appeared in early stages, while in others it was diagnosed at a more advanced age. Although there are differences in the size of the subcohorts, there is a certain tendency with a p-value close to significance (0.1040) that the p.C255Y variant is associated with an earlier onset of hyperuricemia.
In Table 1 there are no statistically significant differences between these recurrent variants, but even so there is a certain tendency for patients with p.C255Y to be in higher CKD stages, whereas 37.5% of p.Q316P carriers continue with an estimated normal glomerular filtration (CKD stage 1) (Fig. 2D).
It is known that the D10C domain is crucial for maintaining the correct three-dimensional structure of the protein; in fact, the group of Ma et al.13 studied the differences in vivo between an alteration within the domain (p.C217 G) and outside it (p.C126R), concluding that when the D10C domain is altered, it has greater deleterious effects at apoptotic level, retention in endoplasmic reticulum, reduced translocation to apical surface and its extracellular release.13,15 This coincides with what was observed in our cohort, in which the patients with the alteration in this domain are in higher stages of CKD (Fig. 2D).
The available data show a higher percentage of patients carrying p.Q316P undergoing RRT and renal transplantation (Table 1). These high proportions may be due to the fact that there are few patients with information on these characteristics, so the percentages may be overestimated.
It is noteworthy that, being the uric acid excretion fraction relevant, this is not usually requested routinely in patients, having information in 8 of the 37 patients.
It is also noteworthy at the clinical level that the type of RRT most commonly used in patients is hemodialysis, used in a total of 9 patients in this cohort (7 of them carry the p.C255Y alteration, and 2, p.Q316P).Peritoneal dialysis is used in 2 patients, one of each type of alteration.
The development of hyperuricemia was the characteristic with the greatest intrafamilial variability in both disorders. Differences were also observed between younger and older relatives, the latter having developed certain age-associated characteristics such as cysts or deterioration of renal function, it being probable that younger individuals develop this symptomatology over time (especially in family F12). These data provide prognostic information associated with these recurrent variants. But this correlation with age is not observed in all cases, for example, F8.S2 presents more severe and earlier symptomatology than F8.S1, despite being younger.
In both subcohorts, guided questioning revealed symptoms such as polydipsia and polyuria, although it seems to be underestimated the increase in urinary volume and increased water intake, due to the low self-perception of these symptoms, which have been constant in the life of these patients.
The main weakness of this study is that, although the 2 subcohorts are made up of patients with comparable ages, the difference in size between them makes it impossible to make a correct comparison in certain characteristics with less available information.
Among the strengths of the study is the clinical observation of 2 recurrent variants in a closed cohort. This provides and enhances knowledge of their characteristics by bringing together a large number of patients carrying the same genetic alteration. Another strong point is the detailed phenotyping of patients, thanks to which valuable clinical information is obtained that allows us to deepen our knowledge of the recurrent variants.
With all this, it is possible to relate the manifestation of characteristics such as the development of hyperuricemia (in some cases even having episodes of gout), renal cysts and deterioration of renal function with alterations in UMOD. Inciding on the recurrent alterations studied, it has been observed that patients carrying p.C255Y tend to present hyperuricemia at earlier ages than carriers of p.Q316P and only carriers of p.C255Y presented episodes of gout. Although no trend was observed in terms of an earlier or later age of onset of advanced CKD, a higher incidence of p.C255Y carriers was observed in stages 3a, 3b and 4. These differences could be due to the fact that the protein structure is altered to different degrees, since the alterations are found in different domains.
The genotype-phenotype correlation of this study is of great prognostic importance as it provides information on how renal pathology could evolve in patients carrying these recurrent variants. As a future perspective, a comparison could be made of other variants in UMOD, and thus learn the particularities that each one may present. Or even extend the study to variants in other genes associated with ADTKD, comparing the phenotype of the patients, and establishing new genotype-phenotype correlations for this disease.
FundingThis study was supported by the Instituto de Salud Carlos III (ISCIII) (REDinREN fund RD016/0009, to M.Á.G.G.; RICORS2040 RD21/0005/0020 and RD24/0004/0010 funded by the European Union-NextGenerationEU, Mecanismo para la Recuperación y la Resiliencia (MRR) to M.Á.G.G.; and projects PI15/01467, PI18/0037 and PI22/00227 co-funded by the European Union to M.Á.G.G.); the Senefro Foundation (SEN2021_2 and SEN2024 to M.Á.G.G.); the Xunta de Galicia/GAIN (Grupos con potencial de crecemento-GPC fund IN607B 2016/020 and IN607B 2023/07 to M. Á.G.G.); and salary support from the Xunta de Galicia (Predoctoral fellowships, to E.S.C.) and from ISCIII through PMP21-000109 (to M.G.M. and E.S.C.).
The authors declare that there is no conflict of interest.
The authors express their gratitude to PKDcore for providing research materials that contribute to expanding knowledge of renal diseases and translational research, and would also like to thank the Art for Dent association for their donations to the research group.
Dr. Alfonso Otero González (CHUOU): 7 patients.
Dr. Jesus Angel Calviño Varela (HULA): 4 patients.
Dr. Alfredo Reparaz Andrade (CHUVI): 3 patients.
Dr. Borja Temes Álvarez (CHUOU): 2 patients.
Dr. Beatriz Millán Díaz (HULA): 2 patients.
Dr. Ana Isabel Díaz Mareque (CHUOU/CHUS): 2 patients.
Dr. Carmen Vázquez Gómez (CHUS): 2 patients.
Dr. Mª Jesús Camba Caride (CHUOU): 2 patients.
Dr. Cándido Díaz Rodríguez (CHUS): one patient.
Dr. Juan José Bravo Lopez (CHUOU): one patient.
Dr. Adela Urisarri Ruiz de Cortaza (CHUS): one patient.
Dr. Alba Rivas Oural (CHUOU): one patient.
Dr. Ana Rodríguez-Carmona de la Torre (CHUAC): one patient.
Dr. Araceli García Pose (HULA): one patient.
Dr. Carmen Cobelo Casas (HULA): one patient.
Dr. Diana Feijoo Piñeiro (CHOP): one patient.
Dr. Elena Iglesias Lamas (CHUOU): one patient.
Dr. Luisa Palomares Solla (CHUVI): one patient.
Dr. Olaia Conde Rivera (CHOP): one patient.
Dr. Miguel Pérez Fontán (CHUAC): one patient.
Dr. María Jesús Castro Vilanova (CHOP): one patient.
Both authors contributed equally to this study.
The names of the GalERH Group researchers who contributed to the UMOD recurrent variant cohort are listed in Appendix A.


AL DÍA

