

Castleman's disease is a rare lymphoproliferative disorder of uncertain etiology. There are 3 histopathological patterns with different clinical manifestations, from localized adenopathies and benign clinical course to systemic manifestations of worse prognosis and high risk of developing into malignant processes.
The following are cases of Castleman's disease with renal involvement treated in the Nephrology Service of the General University Hospital of Ciudad Real, between 2017 and 2024. We describe the clinical evolution and therapeutic management of each of them.
La enfermedad de Castleman es un trastorno linfoproliferativo poco común, de etiología incierta. Existen 3 patrones histopatológicos con manifestaciones clínicas diversas, desde adenopatías localizadas y curso clínico benigno hasta manifestaciones sistémicas de peor pronóstico y alto riesgo de evolucionar hasta procesos malignos.
Presentamos a continuación los casos de enfermedad de Castelman con afectación renal atendidos en el Servicio de Nefrología del Hospital General Universitario de Ciudad Real, entre 2017 y 2024. Describimos la evolución clínica y el manejo terapéutico de cada uno de ellos.
Castleman's disease (CD), or angiofollicular lymphonodular hyperplasia, is a lymphoproliferative disorder of unclear etiology, first described by Benjamin Castleman in 1956.1
It affects women and men with equal frequency and tends to be more common in young adults.
Clinically, it may present as a localized or unicentric entity (UCD), with the appearance of one or more enlarged lymph nodes in a single region of the body and a more indolent course, or as a multicentric involvement (MCD), with generalized lymphadenopathy and a more aggressive clinical expression with systemic involvement (fever, anemia, weight loss, hepatosplenomegaly, increased erythrocyte sedimentation rate and hypergammaglobulinemia). MCD is further subclassified into HHV8-associated MCD, usually observed in immunocompromised patients, and idiopathic MCD (iMCD), which encompasses 4 other different entities: POEMS-associated iCMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder and skin changes), iCMS-TAFRO syndrome (thrombocytopenia, anasarca, myelofibrosis, renal dysfunction and organomegaly), iCMS-LPI (idiopathic plasmacyte lymphadenopathy) and iCMS-NOS or non-specific.2,3
The confirmatory diagnosis is histopathological, with 3 possible variants: hyaline-vascular, the most frequent (80%–90%) that usually corresponds to localized forms of the disease; the plasmocellular variant (10%), generally associated with the multicentric variant and more frequently related to malignant processes, and the mixed variant (2%).4
Regarding treatment, there seems to be consensus on the surgical management of unicentric variants. However, there is no consensus today as to the optimal treatment for the multicentric variety.5
We present a series of 4 cases studied in our center between 2017 and 2023: 3 of them showed hyalinovascular variety, 2 were idiopathic multicentric, with predominantly retroperitoneal adenopathies and the other was unicentric of cervical location. The fourth case was multicentric VHH8 positive with plasmocellular histology. All of them had associated underlying renal disease, most of them of autoimmune etiology.
Reported casesCase 1A 53-year-old man with long-standing hypertension and poor adherence to therapy attended the emergency department for hypertensive crisis (BP 244/144 mmHg). Examination revealed bimalleolar edema. Laboratory tests showed anemia in transfusion range (Hb 6.9 mg/dl) with no evidence of hemolysis (smear without schistocytes, LDH 228 IU/L, reticulocytes 0.79%, haptoglobin 2.8 mg/dl; with normal coagulation, platelets 219,000 mcL). We observed significant deterioration of renal function (Cr 3.8 mg/dl) with subnephrotic proteinuria (protein/creatinine ratio 0.82), without hematuria; the sediment was irrelevant. Reviewing previous studies, without data from 7 years earlier, we found that at the time the patient had normal renal function, without urinary alterations. We performed a study of secondary hypertension, with negative or normal results. In addition, we requested a glomerular protocol with negative antinuclear antibodies (ANA), anti-neutrophil cytoplasm antibodies (ANCA), cryoglobulins and phospholipase A2 receptor antibodies (anti-PLA2R). Immunoglobulins with IgM were 38 IU/ml; the rest, within normal range. There was no monoclonal peak (serum kapppa/lambda ratio 1.17) and complement C3 and C4 were also normal. Proteinogram without alterations. Serology (HCV, HBV, HIV, syphilis), fecal occult blood and tumor markers were also negative. We performed a renal biopsy and obtained the diagnosis of CKD in the context of malignant/accelerated HTN. During admission, we detected a new palpable inguinal adenopathy, so we requested a CT scan, which revealed hepatosplenomegaly and supra- and infradiaphragmatic subcentimetric adenopathies, striking in number, and with uptake in PET-CT, all suggestive of chronic lymphoproliferative syndrome. The biopsy of the inguinal adenopathy described changes compatible with hypervascular Castleman's disease, HHV8 and EBV negative.
We began treatment with cortisone at a dose of 1 mg/kg per day and siltuximab. The evolution was torpid, with the development of pancytopenia and worsening of renal function (Cr 5.6 mg/dl) with an increase in proteinuria to nephrotic range (6.5 protein/creatinine ratio), clinical and analytical nephrotic syndrome. Other associated alterations included severe hypothyroidism and polyclonal hypergammaglobulinemia, not previously present. In this context, acute respiratory failure and decreased level of consciousness occurred, requiring transfer to the ICU, where death finally occurred due to cardiac complications (arrhythmia complications) 2 months after the onset of the hematologic disease.
Case 2A 66-year-old male, who came to the emergency department for asthenia, weight loss and progressive dyspnea of several weeks of evolution. On some occasions, low transient fever. Chest X-ray showed a rounded nodular image in the upper lobe of the right lung. The study continued with CT and confirmed the presence of a well-demarcated right subpleural nodule of 9 × 7 mm, without metabolic activity in PET-CT. In addition, the study described supra- and infradiaphragmatic adenopathies (laterocervical mediastinal, axillary, retroperitoneal periaortic and interaortocaval and bilateral inguinal, the larger ones reaching 12 mm). No organomegaly. During admission, the patient developed a clinical and biochemical nephrotic syndrome (protein/Cr ratio 6.5 g) with microhematuria (sediment 3–5 red blood cells/field) and normal renal function (Cr 0.85 mg/dl). A review of the history showed that previous renal function was normal, without urinary alterations. The immunological study found IgG anticardiolipin (ACA) of 15 U/ml, positive ANCA (not studied by IFA), positive ANA at low titer (1/80), with negative anti-DNA and extractable nuclear antibodies (ENA) and complement consumption (C3 of 57 mg/dl and C4 of 10 mg/dl). In addition, there was anemia in transfusion range (Hb 7.2 mg/dl), without data of hemolysis, mild thrombocytopenia (platelets 138,000 mcL), increased ESR to 41 and polyclonal hypergammaglobulinemia (IgG 1875 mg/dl, IgM 589 mg/dl, IgA 387 mg/dl). We then performed renal biopsy, which revealed membranous nephropathy with focal endocapillary proliferation and C1q deposits, highly suggestive of secondary etiology. PLA 2R negative in biopsy, C4d positive. The analytical study continued with serology (HCV, HBV, HIV, syphilis) and negative tumor markers. Bone marrow biopsy was normal and the puncture of solitary pulmonary nodule showed no evidence of malignancy. Finally, we performed inguinal adenopathy biopsy, in which we found changes compatible with plasmocellular VHH8+ Castleman's disease (negative VHH8 in renal biopsy).
Given the underlying hematological process with renal involvement, we decided, in agreement with the internal medicine department, to initiate treatment with cortisone (prednisone 1 mg/kg per day) and rituximab (375 mg for 8 doses in total), with good clinical and analytical evolution, achieving complete remission after 3 months of treatment. Currently, 4 years after diagnosis and without immunosuppressive maintenance treatment, the patient remains asymptomatic, with preserved renal function and stable adenopathies.
Case 3A 28-year-old woman consulted for neck discomfort in the context of a painless, slow-growing left cervical mass. She also reported night sweats with no other symptoms, dry mouth, no ocular dryness, and ongoing back pain. Examination revealed milk-coffee colored skin lesions. We ordered a body CT which confirmed the presence of bilateral laterocervical adenopathies, the largest 1.5 cm and submaxillary, in the left parotid tail with metabolic activity in PET-CT. No organomegaly. We performed a biopsy of the left cervical adenopathy, showing changes compatible with hyalinovascular Castleman's disease VHH8 and negative EVB-EBER. During the follow-up by internal medicine, an occasional malar rash and ongoing joint pain in the hands and knees appeared. Suspecting a possible associated autoimmune disease (SLE or Sjögren's disease), internal medicine consulted us to extend the study. Laboratory tests showed mild anemia (Hb 10.6 mg/dl) and leukocytopenia (38,000 mcL) with mild lymphopenia. Renal function was normal, with red blood cells and protein in urine (sediment with 5–10 red blood cells/field and protein/creatinine ratio 0.38). Reviewing history, the patient had already presented similar proteinuria and hematuria at least 2 years before. The immunological study revealed positive ANA (titer 1/640) with negative anti-DNA and ENA; lupus anticoagulant positive at high titers (IgM 70, IgG 200); ACA positive (18 U/ml) and hypocomplementemia (C3 of 78 mg/dl and C4 of 6 mg/dl); normal ESR.
Due to the lab results, we started treatment with low-dose corticosteroids (prednisone 0.5 mg/kg per day), siltuximab and acetylsalicylic acid. The patient had a poor clinical and radiological evolution and required admission to the nephrology department for florid nephrotic syndrome (proteinuria up to 6 g, with hypoalbuminemia [2.8 g/dl] and generalized edema). Renal function was normal.
In this context, we started treatment with intravenous corticosteroid boluses (500 mg per 3 doses) together with oral doses at 1 mg/kg per day and scheduled a renal biopsy, with anatomopathological findings compatible with type iv lupus nephropathy and high activity index (11/24). At this point we decided to start treatment with rituximab (375 mg for 8 doses) for a joint approach to lupus nephropathy and Castleman's disease and achieved complete remission of both diseases. The patient has stable hematology lab results and is in complete renal remission. She is on maintenance treatment with prednisone (10 mg/day) and hydroxychloroquine (200 mg/day). A small flare of lupus appeared 2 years after diagnosis, with proteinuria of 1.5 g and minimal reversible deterioration of renal function (Cr 1.45 mg/dl) of probable prerenal etiology. In the sediment: microhematuria with proteinuria by ratio of 1.49. ANA positive (titer 1/320), without having been negative at any time, with negative anti-DNA and positive lupus anticoagulant (IgM 40, IgG 140). We also detected hypocomplementemia (C3 of 70 mg/dl; C4 of 6.5 mg/dl). At that time mycophenolate was associated (500 mg every 12 h), and we increased it to a dose of 1500 mg/day. From the renal point of view there was again remission of the disease (normal renal function, with persistent microhematuria and minimal proteinuria of 0.45 g). However, the systemic symptoms persisted with arthralgias and asthenia, so we agreed with the rheumatology department to associate treatment with belimumab, with good subsequent evolution after 6 months of treatment. Currently, from the nephrological point of view: residual proteinuria around 0.3−0.7 and normal renal function, without adenopathies and with improvement of the systemic symptoms. The patient continues with low-dose prednisone (5 mg/day), mycophenolate (200 mg every 12 h) and belimumab (10 mg/kg monthly), although with poor adherence.
Case 4A 45-year-old woman consulted for a slow-growing breast nodule. The imaging study showed a benign breast lesion, but, incidentally, bilateral axillary adenopathies, without lesions in other areas or organomegaly. No systemic symptomatology at that time. We performed a biopsy of one of the axillary nodes, with a diagnosis of hypervascular Castleman's disease, HHV8 and EBV negative. In the complementary study, the renal function was normal, without urinary alterations. As a result of the immunological protocol, ANA was positive at low titer (1/160), with positive anti-DNA, negative ENA and polyclonal hypergammaglobulinemia (IgG 1715 mg/dl, IgM 679 mg/dl, IgA 407 mg/dl). No complement consumption and negative ANCA, ACA and lupus anticoagulant. Bone marrow biopsy, without evidence of infiltration. In the directed interview, the patient reported chest skin lesions on some occasions, with malar rash, oral aphthous ulcers and generalized arthralgias, which she no longer presents. Treatment with hydroxychloroquine (200 mg/day) and follow-up was indicated. Two years after CD diagnosis, the patient began suffering clinical and biochemical nephrotic syndrome with proteinuria of 8 g, bimalleolar edema and hypoalbuminemia of 2.3 mg/dl and microhematuria of 5 red blood cells/field in sediment. Renal function was normal. In the immunological analysis, ANA positivity persisted, now at a titer of 1/640 with positive anti-DNA and positive anti-ENA (SS-A: ++, Ro 52: ++, SS-B: +, nucleosomes +, histones +, AMA M2: +++). She had persistent polyclonal hypergammaglobulinemia and complement consumption (C3 of 48 mg/dl and C4 of 7 mg/dl). During this time, the adenopathies remained stable. We then decided to perform a renal biopsy, that showed histology of type iv lupus nephropathy. We maintained treatment with antimalarials (hydroxychloroquine at 200 mg/day) and started treatment with intravenous corticosteroids (3 boluses of 500 mg) oral dose of 1 mg/kg per day in a descending pattern and mycophenolate (250 mg every 12 h), achieving good clinical and analytical response. Currently the patient is in complete remission, with preserved renal function and stable adenopathies. She maintains treatment with prednisone 2.5 mg every other day with mycophenolate in a descending regimen of 250 mg per day and antimalarials. The rheumatology service raised the possibility of associating belimumab before the complete withdrawal of mycophenolate.
DiscussionWe studied 4 patients diagnosed with CD between 2017 and 2024 in our clinic.
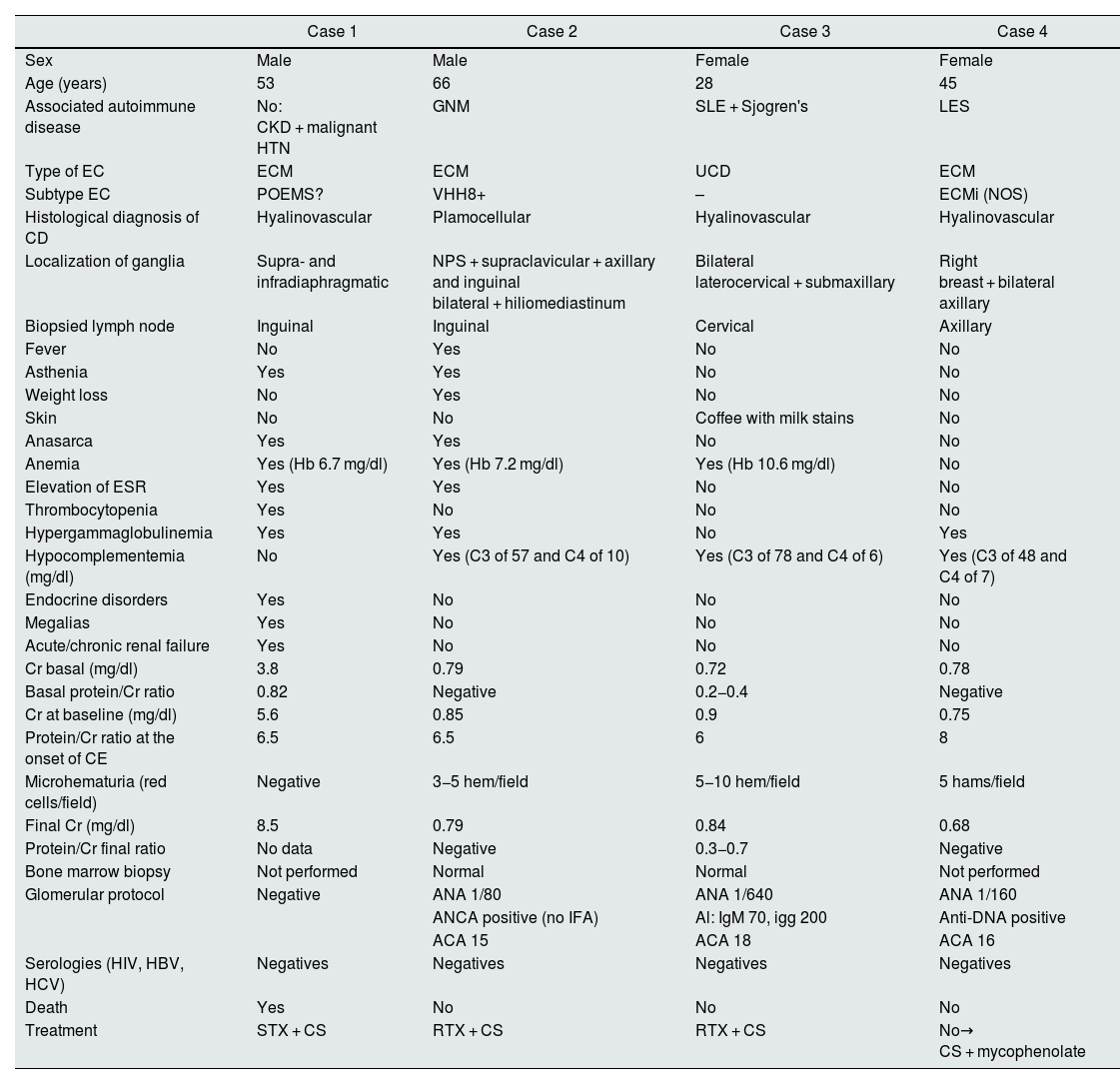
The disease presented with equal frequency in men and women, in contrast to reports in the medical literature.1 The mean age of presentation was 48 years and the most frequent location for the presentation of adenopathies was retroperitoneal. Only in the case of UCD were they identified in the cervical area. Table 1 shows the epidemiological data, clinical characteristics and analytical results of all patients.
Epidemiological data, clinical characteristics and analytical results of patients with Castleman's disease.
| Case 1 | Case 2 | Case 3 | Case 4 | |
|---|---|---|---|---|
| Sex | Male | Male | Female | Female |
| Age (years) | 53 | 66 | 28 | 45 |
| Associated autoimmune disease | No: CKD + malignant HTN | GNM | SLE + Sjogren's | LES |
| Type of EC | ECM | ECM | UCD | ECM |
| Subtype EC | POEMS? | VHH8+ | – | ECMi (NOS) |
| Histological diagnosis of CD | Hyalinovascular | Plamocellular | Hyalinovascular | Hyalinovascular |
| Localization of ganglia | Supra- and infradiaphragmatic | NPS + supraclavicular + axillary and inguinal bilateral + hiliomediastinum | Bilateral laterocervical + submaxillary | Right breast + bilateral axillary |
| Biopsied lymph node | Inguinal | Inguinal | Cervical | Axillary |
| Fever | No | Yes | No | No |
| Asthenia | Yes | Yes | No | No |
| Weight loss | No | Yes | No | No |
| Skin | No | No | Coffee with milk stains | No |
| Anasarca | Yes | Yes | No | No |
| Anemia | Yes (Hb 6.7 mg/dl) | Yes (Hb 7.2 mg/dl) | Yes (Hb 10.6 mg/dl) | No |
| Elevation of ESR | Yes | Yes | No | No |
| Thrombocytopenia | Yes | No | No | No |
| Hypergammaglobulinemia | Yes | Yes | No | Yes |
| Hypocomplementemia (mg/dl) | No | Yes (C3 of 57 and C4 of 10) | Yes (C3 of 78 and C4 of 6) | Yes (C3 of 48 and C4 of 7) |
| Endocrine disorders | Yes | No | No | No |
| Megalias | Yes | No | No | No |
| Acute/chronic renal failure | Yes | No | No | No |
| Cr basal (mg/dl) | 3.8 | 0.79 | 0.72 | 0.78 |
| Basal protein/Cr ratio | 0.82 | Negative | 0.2−0.4 | Negative |
| Cr at baseline (mg/dl) | 5.6 | 0.85 | 0.9 | 0.75 |
| Protein/Cr ratio at the onset of CE | 6.5 | 6.5 | 6 | 8 |
| Microhematuria (red cells/field) | Negative | 3−5 hem/field | 5−10 hem/field | 5 hams/field |
| Final Cr (mg/dl) | 8.5 | 0.79 | 0.84 | 0.68 |
| Protein/Cr final ratio | No data | Negative | 0.3−0.7 | Negative |
| Bone marrow biopsy | Not performed | Normal | Normal | Not performed |
| Glomerular protocol | Negative | ANA 1/80 | ANA 1/640 | ANA 1/160 |
| ANCA positive (no IFA) | Al: IgM 70, igg 200 | Anti-DNA positive | ||
| ACA 15 | ACA 18 | ACA 16 | ||
| Serologies (HIV, HBV, HCV) | Negatives | Negatives | Negatives | Negatives |
| Death | Yes | No | No | No |
| Treatment | STX + CS | RTX + CS | RTX + CS | No→ CS + mycophenolate |
CKD: chronic kidney disease; HT: arterial hypertension; MGN: membranous glomerulonephritis; SLE: systemic lupus erythematosus; CD: Castleman's disease; MCD: multicentric Castleman's disease; UCD: unicentric Castleman's disease; POEMS: polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes; HHV8: human herpes virus 8; ECMi (NOS): idiopathic Castleman's disease (nonspecific); SPN: solitary pulmonary nodule; ESR: glomerular sedimentation rate; Cr: creatinine; ANA: antinuclear antibodies; ANCA: anti-neutrophil cytoplasmic antibodies; DNA: deoxyribonucleic acid; ACA: anticardiolipin antibodies; HIV: human immunodeficiency virus; HBV: hepatitis B virus; HCV: hepatitis C virus; STX: siltuximab; RTX: rituximab; CS: corticosteroids.
According to the literature, the most common form of presentation is localized,2,5,6 but we diagnosed 3 of the 4 cases with MCD, 2 idiopathic and one associated with HHV8. The most frequent variant was hyalinovascular, in 75% of the cases, and only one case, the multicentric one associated with VHH8, corresponded to the plasmocellular variant. According to the literature, CD associated with HHV8 is related to HIV infection in more than 40% of cases.7 In our patient, serologies were negative. Fig. 1 shows the anatomopathological description of the lymph nodes.
 Panoramic view of part of the lymph node with central medullary and cortical follicular pattern with dilated sinusoids. B) At higher magnification, hypoplastic-atrophic lymphoid follicles, in the germinal centers hyalinized wall vessels and surrounded by concentric mantle lymphoid cells (“onion layer arrangement”). C) In interfollicular areas there is proliferation of mature looking plasmacytoid cells. D) There are no IHC images, but the result was negative for HHV8 and EBV-EBER, as well as polytypic expression of light chains. Positive expression of CD30 limited to follicular blasts. Case 2: A) Panoramic view. Lymph node excisional biopsy with central medullary and lymph node cortex with effacement of the usual lymph node architecture. B) Presence of clusters of blasts (inset). C) Marked lymphoplasmacytosis (plasma marked with circles). D) IHC shows positivity in the blasts for HHV-8. Case 3: A) Panoramic view. Excisional lymph node biopsy with follicular pattern. B and C) Higher magnification, hypoplastic-atrophic lymphoid follicles, in germinal centers hyalinized wall vessels and surrounded by mantle lymphoid cells concentrically (“onion layer arrangement”). D) IHC shows marked expression of CD23 and CD21 in germinal centers. Case 4: No imaging.")
Pathological description of lymph nodes. Case 1: A) Panoramic view of part of the lymph node with central medullary and cortical follicular pattern with dilated sinusoids. B) At higher magnification, hypoplastic-atrophic lymphoid follicles, in the germinal centers hyalinized wall vessels and surrounded by concentric mantle lymphoid cells (“onion layer arrangement”). C) In interfollicular areas there is proliferation of mature looking plasmacytoid cells. D) There are no IHC images, but the result was negative for HHV8 and EBV-EBER, as well as polytypic expression of light chains. Positive expression of CD30 limited to follicular blasts. Case 2: A) Panoramic view. Lymph node excisional biopsy with central medullary and lymph node cortex with effacement of the usual lymph node architecture. B) Presence of clusters of blasts (inset). C) Marked lymphoplasmacytosis (plasma marked with circles). D) IHC shows positivity in the blasts for HHV-8. Case 3: A) Panoramic view. Excisional lymph node biopsy with follicular pattern. B and C) Higher magnification, hypoplastic-atrophic lymphoid follicles, in germinal centers hyalinized wall vessels and surrounded by mantle lymphoid cells concentrically (“onion layer arrangement”). D) IHC shows marked expression of CD23 and CD21 in germinal centers. Case 4: No imaging.
As for the clinical manifestations, although published studies report that multicentric involvement usually presents a more aggressive behavior with systemic involvement,8 in our experience only the VHH8-positive multicentric type with plasmocellular variant manifested from the beginning, with fever, anemia, asthenia and weight loss. These alterations appear to be associated with increased IL-6 secretion and can often be complicated by the development of amyloidosis, infections and malignancies, particularly Kaposi's sarcoma or lymphoma.5,7 In the rest of the patients, the reason for consultation was palpable adenopathies or their incidental finding in studies for other causes. We could consider here that the aggressiveness of the clinical picture, rather than the anatomical classification, could be determined by the histological presentation and correspond to the most severe cases, with the plasmocellular variant. Some studies already propose this hypothesis.7,10
Although none of the cases of idiopathic SCD included in the study were subclassified according to the entities present in the literature (POEMS, TAFIRO, LPI, NOS), we observed that one of our patients, in addition to the systemic manifestations mentioned above, presented organomegaly, thrombocytopenia and severe endocrine alterations, which led us to suspect a posteriori the POEMS variant. This was the only case of CD with renal involvement prior to diagnosis and without associated underlying autoimmune disease. In all the other patients (cases 2, 3 and 4), we found an immunologic substrate, such as SLE and secondary MGN, diagnosed by renal biopsy. The doubt we have at this point is whether we are dealing with autoimmune manifestations of CD or with autoimmune processes whose lymph node histology resembles that of SCD. The 2 cases with a diagnosis of SLE (cases 3 and 4) are the ones that generate the greatest uncertainty for us, since, according to the expert consensus for the integrated diagnosis of idiopathic multicentric CD, between 15% and 30% of patients with lupus show a lymph node histopathology similar to that of SCID.9 We did not find in the literature any studies that go deeper into this aspect, apart from the mention of diseases such as SLE, rheumatoid arthritis, autoimmune lymphoproliferative syndrome and Still's disease as possible exclusion criteria for CD.9,11 In our case series, the diagnosis of CD was prior to that of SLE, with a time gap of months or even years until the onset of renal disease, so they were considered as coexisting entities, although with a related immunological substrate to be considered at the time of treatment.
Similarly, reviewing the renal involvement of CD, very few articles provide a detailed description in this regard, hence the interest of our work. Most of the studies reviewed briefly describe a clinical manifestation marked by acute renal failure with increased plasma creatinine and associated nephrotic syndrome, but do not provide further data on analytical values and chronological evolution.2–4,9 In our case series, we have collected these aspects. As seen in Table 1, only case 1 presented deterioration of renal function prior to the diagnosis of CD, although we do not consider its etiology to be related to the hematologic disease itself. The rest of the cases, and unlike previous reports in the literature, preserved renal function throughout the acute process. All of them developed a clinical and biochemical nephrotic syndrome, with marked proteinuria above 6 g, and alterations in the urinary sediment in the form of microhematuria, something not previously described. Regarding autoimmunity, ANAs were present in all patients, with different titers, although their nonspecific nature prevents us from drawing conclusions in this regard. In all these cases, there was complement consumption, something that is already contemplated in CD without renal involvement.9,11 The presence of ACA in most of the patients did draw our attention.
Reviewing the literature, we did not find any mention that relates these autoantibodies to CD, so perhaps it would be interesting to take this into account for future studies.
Finally, it is worth mentioning the anatomical pathologic findings confirmed by kidney biopsy performed in each of our patients. Given that renal involvement in most cases is very infrequent, we also did not find large studies with a clear established pattern of histological changes. Reviewing publications of isolated clinical cases and small series of cases, it seems that the 2 lesions most frequently observed from the histopathological point of view in patients with CD and renal involvement were thrombotic microangiopathy-like (TMA-like) and secondary amyloidosis.12–14 However, as seen in Fig. 2, the anatomopathological description in our sample differs from other publications to date. Most cases describe a thickening of the basement membrane, with “moth-eaten” images and mesangial expansion, with endocapillary proliferation at the expense of mononuclear cells and some neutrophils. In the interstitial compartment, we describe mild fibrosis, with minimal tubular necrosis and without inflammatory infiltrate, or with minimal presence of mononuclear cells. In most cases we observed no vascular damage. Case 1 did present vascular damage, with intimal hyperplasia associated with malignant hypertension, but at no time was the relationship with CD considered. In all of them, Congo red staining was negative and direct immunofluorescence was consistent with the renal diagnosis, as is visible in the images.
Regarding the treatment of the localized form, there is a uniform criterion in the literature that it should be treated surgically.4,10,15 However, in our case, in the only patient classified as a unicentric variant (case 3), we chose medical management and follow-up.
Regarding the treatment of multicentric variants, there seems not to be a consensus. Variable results have been obtained with chemotherapy alone, in combination with steroids and also with radiotherapy.14,16,17 In our case, patient number 1 was treated with siltuximab, perhaps because the renal involvement was attributed to a non-autoimmune etiology from the beginning. In the other two patients with multicentric variants, the use of rituximab was preferred as a joint approach of both processes. Most of them also received corticosteroid treatment from the beginning.
To date, the three patients who remain alive are asymptomatic, including the plasmocellular variant, and without renal autoimmune disease activity. Due to the potentially malignant nature of the multicentric or plasmocellular form, patients should be monitored and evaluated periodically, with no description to date of the follow-up time necessary to consider cure. Long-term development of complications such as Kaposi's sarcoma or non-Hodgkin's lymphoma cannot be ruled out.
ConclusionCD is a rare benign lymphoproliferative disorder, presenting in the lymph nodes as follicular lymphoid hyperplasia.
Among its clinical manifestations, renal involvement is very rare; it appears in most cases as clinical and biochemical nephrotic syndrome with or without renal function impairment.
No specific autoimmunity is known to be associated with this entity. Our results indicate that anticardiolipin antibodies could be present when there are associated autoimmune processes, although only larger studies could establish a direct relationship. We also did not find a consistent histological pattern in the renal biopsy. The thickening of the basement membrane, with mesangial expansion and endocapillary proliferation, are the most repeated findings in our study. We did not observe vascular damage or positivity for Congo red.
There is a clear need for more studies, as well as sharing and analyzing each of the cases diagnosed in each center, to better understand the pathogenesis, pathophysiology and natural evolution of this disease, since only in this way will diagnostic and therapeutic options improve.



