





La enfermedad relacionada con IgG4 es un trastorno sistémico autoinmune de etiopatogenia desconocida con características histopatológicas propias y aumento de IgG4 en suero y tejidos afectos. Se presenta de modo predominante en varones mayores de 60 años y la manifestación renal más frecuente es la nefritis tubulointersticial, que aparece como una insuficiencia renal aguda o crónica o como nódulos o masas renales radiológicas, o ambas. En algunos casos se acompaña de alteraciones urinarias, lo que obliga a descartar un proceso glomerular, fundamentalmente una nefropatía membranosa. La mayoría de las veces se asocia con lesiones extrarrenales esclerosantes en varios órganos o glándulas, sobre todo, sialoadenitis, linfadenopatía o pancreatitis autoinmune. Serológicamente se detectan niveles elevados de IgG total e IgG4 séricas en la mayoría de los casos, frecuentemente aumento de IgE e hipocomplementemia y a veces eosinofilia. La biopsia muestra un infiltrado intersticial inflamatorio rico en células plasmáticas que secretan IgG4, fibrosis intersticial expansiva y depósitos de inmunocomplejos en la membrana basal tubular. La respuesta a esteroides suele ser eficaz y característica de la enfermedad, aunque la recidiva es frecuente y obliga a un tratamiento de mantenimiento. En caso de corticorresistencia o corticodependencia, se plantean otras alternativas inmunosupresoras, aunque la más aceptada es rituximab.
Conceptos clave
1. Es una enfermedad sistémica de etiología desconocida en la que hay un aumento de IgG4 en suero y en los tejidos afectos de significado incierto.
2. A diferencia de otras enfermedades autoinmunes, es más frecuente en varones de edad avanzada.
3. Se suele presentar como lesiones esclerosantes en varios órganos o glándulas.
4. Puede afectar a cualquier compartimento renal, aunque generalmente provoca nefritis intersticial con disfunción renal aguda o crónica.
5. La aparición de alteraciones urinarias obliga a descartar una NM asociada.
6. El aumento de IgG e IgG4 no es exclusivo de la enfermedad y suele acompañarse de hipocomplementemia, eosinofilia y una imagen radiológica de pseudotumor inflamatorio.
7. La biopsia es clave en el diagnóstico y se observa un denso infiltrado linfoplasmocitario rico en IgG4 con un patrón de fibrosis característico y depósito de inmunocompejos en la membrana basal tubular.
8. La respuesta a esteroides es la regla.
INTRODUCCIÓN
La IgG4 ha unificado un considerable número de patologías previamente no relacionadas. Estas enfermedades forman un grupo heterogéneo de trastornos autoinmunes inflamatorios crónicos, sistémicos y multiorgánicos, con características propias, clínicas, serológicas, radiológicas, histopatológicas y de respuesta al tratamiento.
La enfermedad relacionada con IgG4 (IgG4 related disease o IgG4-RD) se debe sospechar en pacientes con pancreatitis idiopática, colangitis esclerosante, fibrosis retroperitoneal, afectación de glándulas salivales o lacrimales, o con un pseudotumor orbitario o proptosis del globo ocular, solos o en combinación, con síntomas alérgicos y niveles séricos elevados de IgG4.
A pesar de los diferentes tejidos implicados, sus características histopatológicas tienen sorprendentes similitudes: un infiltrado linfoplasmocitario denso, un patrón de fibrosis típico, eosinofilia y el acúmulo de células plasmáticas secretoras de IgG4 en los tejidos afectados1.
HISTORIA
En 1961 se describió el caso de una pancreatitis esclerosante con hipergammaglobulinemia2, pero no fue hasta 1995 cuando se pudo demostrar la naturaleza autoinmune de esta entidad3.
En 2001 se estableció la relación entre la IgG4 y la pancreatitis esclerosante al encontrarse un aumento de niveles séricos de IgG44. En 2002 se demostró la infiltración tisular por células plasmáticas secretoras de IgG4 en el páncreas de pacientes con pancreatitis autoinmune5 y en 2003 en otros tejidos, confirmando el carácter sistémico de la enfermedad, que fue etiquetada de «enfermedad sistémica relacionada con IgG4»6.
Las primeras referencias al riñón datan de 2004 en Japón, donde se publicó un caso de nefritis tubulointersticial (NTI) no granulomatosa en un paciente con pancreatitis autoinmune relacionada con IgG4 y una NTI en la biopsia de unos nódulos renales en un paciente con la misma patología7,8. Pocos años después se hizo la primera descripción de una nefropatía membranosa (NM) asociada a NTI relacionada con IgG4 (NTI-IgG4)9.
En octubre de 2011 se propuso el término «IgG4-related kidney disease» en el simposio internacional de Boston, la primera conferencia sobre IgG4-RD, y en 2012 se gestó el primer documento de consenso sobre la enfermedad10.
LA IgG4
Las subclases de IgG tienen diferencias estructurales en distintos niveles de la molécula que son en gran parte responsables de las distintas propiedades biológicas y de que determinados Ag provoquen una respuesta restringida a una subclase determinada.
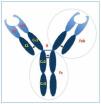
La molécula de IgG4 (figura 1) es la subclase de IgG de menor tamaño y la más escasa en suero (< 5 % de IgG total)11. Una exposición crónica o repetida al Ag conlleva títulos de IgG4 persistentemente elevados y estables12.

Figura 1.Molécula de IgG4. §: puentes disulfuro entre las cadenas (fuerzas no covalentes); B (bisagra): en las cadenas H, muy flexible, permite adquirir distintos ángulos entre las regiones V y C, y entre los brazos de la Ig; C: regiones carboxi-terminales que poseen muy poca variación en su secuencia, responsables de las funciones efectoras de la IgG; Fab: fragmento de unión al Ag; Fc (fragmento cristalizable): asegura una respuesta inmune adecuada al unirse a receptores celulares y proteínas del complemento; H: dos cadenas pesadas idénticas; L: dos cadenas ligeras idénticas; V: regiones amino-terminales con una gran variabilidad en su secuencia de aminoácidos, implicadas en el reconocimiento y unión del Ag.
La IgG4 tiene poca flexibilidad en su región bisagra, lo que limita sus funciones efectoras de unión al complemento y a receptores Fc que expresan numerosas células implicadas en la fagocitosis y liberación de mediadores. En algunas circunstancias puede unirse a la porción Fc de otras IgG e incluso a otras moléculas de IgG411,12. Pequeñas diferencias en determinadas secuencias de aminoácidos le confieren una baja afinidad por los receptores de C1q incapacitándola para activar el complemento13.
Los Ac IgG forman inmunocomplejos por su capacidad para unir dos moléculas de Ag. Sin embargo, las moléculas de IgG4 por su capacidad de intercambiar brazos Fab con otras moléculas de IgG4 y otros isotipos de Ac se transforman en Ac biespecíficos pero funcionalmente monovalentes, formando inmunocomplejos ineficaces, incapaces de precipitar y bloqueando la unión del Ag a IgG1, que es más patógena14,15.
PATOGENIA
No se sabe con certeza el papel de IgG4 en la patogénesis de las IgG4-RD. Se ha especulado sobre su carácter patógeno, aunque hoy se cree que su sobreexpresión es una consecuencia de un estímulo inflamatorio desconocido en personas genéticamente predispuestas16.
La producción de IgG4 (y también de IgE) se controla principalmente por las células T helper 2 (Th2)12,13. En los órganos afectados por las IgG4-RD se ha demostrado el predominio de células Th2 y la posterior activación de células T reguladoras (Treg)17. Se ha probado la sobreexpresión de IL-4, IL-10 y del factor de crecimiento transformante β en las NTI-IgG4 en comparación con otros tipos de NTI, lo que indica que Th2 y Treg parecen jugar un papel central en las NTI-IgG418. Estas citocinas contribuyen a la eosinofilia, al aumento de las concentraciones séricas de IgG4 e IgE, y a la progresión de la fibrosis. Se produciría una infiltración masiva de células inflamatorias en distintos órganos, tumefacción, expansión de la fibrosis y disfunción orgánica como consecuencia de la inflamación tisular y el depósito de inmunocomplejos1.
EPIDEMIOLOGÍA
Es más frecuente en varones (75-85 %) de 60-70 años (rango 20-81)19-23, salvo en casos limitados a cabeza y cuello, donde no hay diferencias por sexo24.
Existen escasos estudios genéticos y limitados a la población asiática. Se desconoce la causa de la mayor prevalencia en Japón y de la diferencia con el resto de los países occidentales. Es posible que haya causas genéticas, pero parece que la IgG4-RD está infradiagnosticada.
PRESENTACIÓN CLÍNICA
La enfermedad multiorgánica es más frecuente (60-90 %) que la afectación aislada de un solo órgano y las diferentes manifestaciones de la IgG4-RD no son necesariamente coincidentes en el tiempo, pudiendo aparecer meses e incluso años después de la presentación, llegando al diagnóstico incluso tras revisar la histopatología previa24.
La pancreatitis autoimmune tipo 1 es el prototipo de IgG4-RD y la forma extrapancreática más frecuente es la colangitis esclerosante. Puede afectar prácticamente a todo el organismo: hipófisis, glándulas salivales y lacrimales (la enfermedad de Miculicz afecta a las glándulas lacrimal, parótida y submandibular), ojos, órbitas, sistema nervioso central y periférico (paquimeningitis, neuritis infraorbitaria), tiroides (tiroiditis de Riedel y la variante fibrosa de la tiroiditis de Hashimoto), ganglios linfáticos, mediastino, pulmón, pleura y pericardio, mamas, estómago, hígado y vías biliares, retroperitoneo (periaortitis, fibrosis retroperitoneal), riñón, próstata y la piel (pseudolinfoma cutáneo)25.
La IgG4-RD puede presentarse como:
— Sintomática: fiebre, fatiga, anorexia o dolor abdominal, frecuentemente con linfadenopatía que a veces es el signo inicial, y datos de rinitis alérgica o asma bronquial hasta en un 40 % de los pacientes26.
— Asintomática: diagnóstico incidental por alteraciones de laboratorio y/o de imagen por la presencia de una masa (pseudotumor orbitario, masa renal o pancreática, nódulos pulmonares) o por el agrandamiento difuso de un órgano (pancreatitis) o glándula27.
PRESENTACIÓN RENAL
En el riñón la IgG4-RD afecta fundamentalmente al intersticio (NTI-IgG4)19, puede afectar al glomérulo, generalmente en forma de NM, aunque se han descrito casos de nefropatía IgA, vasculitis IgA, glomerulonefritis membranoproliferativa y glomerulonefritis proliferativa endocapilar con semilunas19,23, y por último puede afectar a los vasos (arteritis relacionada con IgG4)28.
La NTI-IgG4 es la afectación parenquimatosa renal más frecuente. Puede encontrarse en el 15 % de los pacientes con IgG4-RD, el 96 % de los cuales tiene afectación de otros tejidos (sialoadenitis 83 %, linfadenopatía 44 %, pancreatitis 39 %, dacrioadenitis 30 %, lesiones pulmonares 26 %)19.
Puede aparecer como una insuficiencia renal aguda o crónica progresiva (77 %) con afectación extrarrenal (83 %), generalmente multiorgánica. Globalmente se ha constatado proteinuria < 1 g/24 horas en el 70 % de los casos y microhematuria en el 22 %20.
El 78 % tiene alteraciones radiológicas, generalmente bilaterales y múltiples (75 % como masa renal y 25 % como nefromegalia). Se comportan como un pseudotumor inflamatorio que no pocas veces ha derivado en una nefrectomía y se corresponde histológicamente con una NTI-IgG423. Se ha descrito también en injertos renales29.
La NM relacionada con IgG4 (NM-IgG4) es la glomerulopatía más frecuente y está presente en el 7-10 % de los pacientes con NTI-IgG419,22,23,30.
La mayoría de los casos de NM-IgG4 se acompaña de NTI9,19,21,22 y en los casos en los que la NTI está ausente se detectan otros datos de IgG4-RD30,31. Hasta el 75 % de los pacientes con NMIgG4 presentan afectación extrarrenal. Sin embargo, los casos de IgG4-RD en que la lesión glomerular es la única lesión renal (sin NTI) no se definen como IgG4-RD, aunque esta debería descartarse en casos de NM e historia clínica sugestiva de IgG4-RD21,30.
La proteinuria suele ser de rango nefrótico y tienen microhematuria, la mayoría con insuficiencia renal31. En todos los casos los Ac anti-PLA2R son negativos30. Sin embargo, se ha demostrado una NM-IgG4 tres años después del diagnóstico de una NM idiopática, lo que puede sugerir una relación fisiopatológica entre ambas (reacciones inmunomediadas por Th2)32 e incluso hay un caso descrito de NM no relacionada con IgG4 con Ac IgG3 circulantes frente a superóxido dismutasa 2 en una paciente con IgG4-RD remitida previamente31.
Además de la afectación parenquimatosa renal, la IgG4-RD puede presentarse como una uropatía obstructiva, no solo en algunos pseudotumores renales, sino también en otras entidades como la fibrosis retroperitoneal por IgG433, la pielitis crónica esclerosante, que es una masa inflamatoria que afecta a la pelvis renal y patológicamente es similar a la NIT-IgG434, el pseudotumor ureteral inflamatorio35 o la prostatitis por IgG436.
DATOS DE LABORATORIO
— Algunos pacientes tienen microhematuria y menos de la mitad proteinuria, generalmente leve y rara vez de rango nefrótico19,21, salvo que haya una glomerulopatía asociada31.
— La función renal puede ser normal o desarrollar insuficiencia renal, aguda o lentamente progresiva19-23.
— También podemos encontrar una hipergammaglobulinemia policlonal, eosinofilia (33-48 %), FR positivo y PCR ligeramente elevada.
— AutoAc: ANA a título bajo (30 %), el resto, negativos (anti-PLA2R, anti-DNAn, anti-SS-A, anti-SS-B, anti-Sm, anti-RNP, ANCA, crioglobulinas, y proteína M)19-23.
— Nefelometría: hipocomplementemia C3 y/o C4 (50-78 %), niveles séricos elevados de IgE (70 %)19-23,25 e IgG total (90 %)21. Los niveles de IgG4 sérica están elevados en la mayoría de los pacientes. El aumento de IgG4 en suero (> 135 mg/dl) es el marcador diagnóstico más importante y ocurre en el 80 % de los casos1,21, aunque no es exclusivo de la IgG4-RD y sus resultados deben ser interpretados con precaución37. Sus niveles no se relacionan con el grado de actividad, pero son útiles para el seguimiento38.
— El índice IgG4/IgG en orina es un marcador del deterioro de la barrera de selectividad y representa el elevado aclaramiento renal de IgG4. Pues bien, se ha observado que es significativamente mayor en la NM idiopática que en la NM-IgG439.
— El número de plasmoblastos IgG4 circulantes analizados por citometría de flujo aumenta cuando la IgG4-RD está activa, incluso en pacientes con concentraciones normales de IgG4 sérica, por lo que puede ser un biomarcador potencialmente útil para el diagnóstico, evaluar la respuesta al tratamiento e indicar un tratamiento precoz en las recidivas40.
ESTUDIOS DE IMAGEN
La presencia de alteraciones renales en los estudios de imagen es frecuente y una característica diferencial de otros tipos de NTI19-23.
Ecografía abdominal: lesiones hipoecogénicas traducen la inflamación del parénquima41.
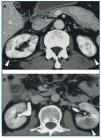
Tomografía computarizada (TC) con contraste (figura 2): las lesiones parenquimatosas son generalmente bilaterales, múltiples, y afectan de manera predominante a la corteza renal. En el 65 % se presentan como pequeños nódulos corticales periféricos redondeados, hipovasculares, de baja intensidad, invadiendo la cápsula pero sin malignidad en el examen histológico21. Otras veces se ven lesiones cuneiformes o difusas irregulares o grandes masas solitarias (pseudotumor inflamatorio)19-21 o incluso una masa quística42. La pielitis se insinúa como un engrosamiento difuso de la pared de la pelvis con una superficie lisa intraluminal21. Puede también manifestarse como una hidronefrosis22,27.
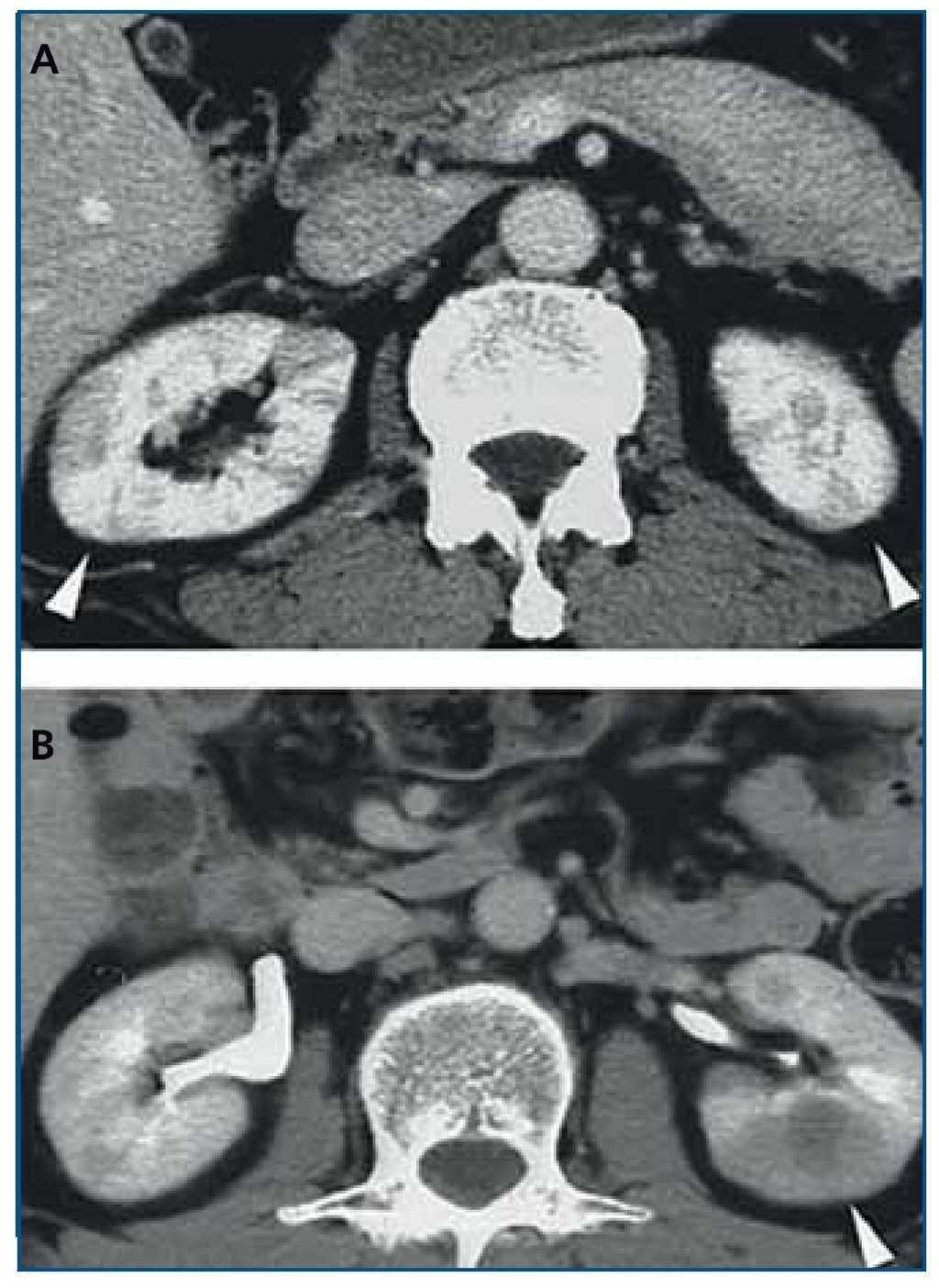
Figura 2. Tomografía computarizada con contraste. A) Múltiples lesiones cuneiformes de baja intensidad en ambas corticales renales; B) Pseudotumor inflamatorio en el riñón izquierdo. Adaptado con permiso de: Macmillan Publishers Ltd: Kidney Int19, copyright 2010.
Resonancia magnética nuclear (RNM): las imágenes pseudo-tumorales son bilaterales (84 %), múltiples (93 %), parenquimatosas (87 %), iso-hipointensas, y se ven mejor incluyendo imágenes en difusión en la RNM convencional41,43.
El renal rim sign es un signo radiológico (TC o RNM) que representa el proceso inflamatorio extendiéndose al tejido adiposo extrarrenal, como un borde de tejido blando alrededor de los riñones44.
La gammagrafía con galio y la tomografía por emisión de positrones son también útiles para identificar lesiones renales y extrarrenales19-21.
HISTOPATOLOGÍA
El análisis histopatológico es la piedra angular en el diagnóstico de la IgG4-RD. Se describe una tumefacción pseudotumoral compuesta por un denso infiltrado linfoplasmocitario rico en células plasmáticas IgG4 y linfocitos T CD4, y un grado variable de fibrosis con un patrón de disposición celular (fibroblastos, células inflamatorias) «estoriforme» (enmarañado e irregularmente verticilado), flebitis obliterante y un infiltrado leve-moderado de eosinófilos24.
El número de células plasmáticas IgG4/campo de gran aumento (HPF) varía poco de un tejido a otro. Generalmente, el mínimo para hacer el diagnóstico en la mayoría de los tejidos es de 30 a 50 células plasmáticas IgG4/HPF; sin embargo, en algunos órganos o tejidos con solo 10 células plasmáticas IgG4/HPF puede ser suficiente10.
Las características microscópicas e inmunohistoquímicas son muy similares independientemente del órgano o tejido implicado, pero en una fase tardía de la enfermedad el diagnóstico es más difícil debido al menor número de células plasmáticas y al predominio de la fibrosis. En este sentido, el patrón de la fibrosis y la proporción de IgG4/IgG total aporta información fundamental.
En el riñón, la presencia de numerosas células plasmáticas IgG4 es esencial, pero no suficiente, para el diagnóstico45. La infiltración de > 10 células plasmáticas IgG4/HPF (biopsia con aguja)23 y la relación IgG4/IgG > 40 %32,67 son muy características, aunque no exclusivas. La infiltración por IgG4 se ha detectado en otras enfermedades renales como la glomerulonefritis necrotizante pauciinmune con semilunas y en menor intensidad en la NTI autoinmune y por drogas20,46.
Desde el punto de vista macroscópico, el parénquima renal es blanquecino, de consistencia firme, homogénea y sólida41, y las lesiones de la pelvis renal muestran un engrosamiento de la pared34.
Microscópicamente son lesiones focales o difusas, que afectan tanto a la corteza renal como a la médula, bien delimitadas y diferenciadas del parénquima sano, por lo que a veces no se identifican en las biopsias con aguja22,23,47.
Se diferencian las siguientes lesiones.
Lesiones tubulointersticiales (NTI-IgG4)
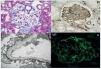
Microscopio óptico (MO): denso infiltrado linfoplasmocitario rico en células plasmáticas IgG4 (sin atipias) y fibrosis que se extiende desde el parénquima a la pelvis renal y al tejido adiposo perirrenal10,19,22,25,47 (figura 3).
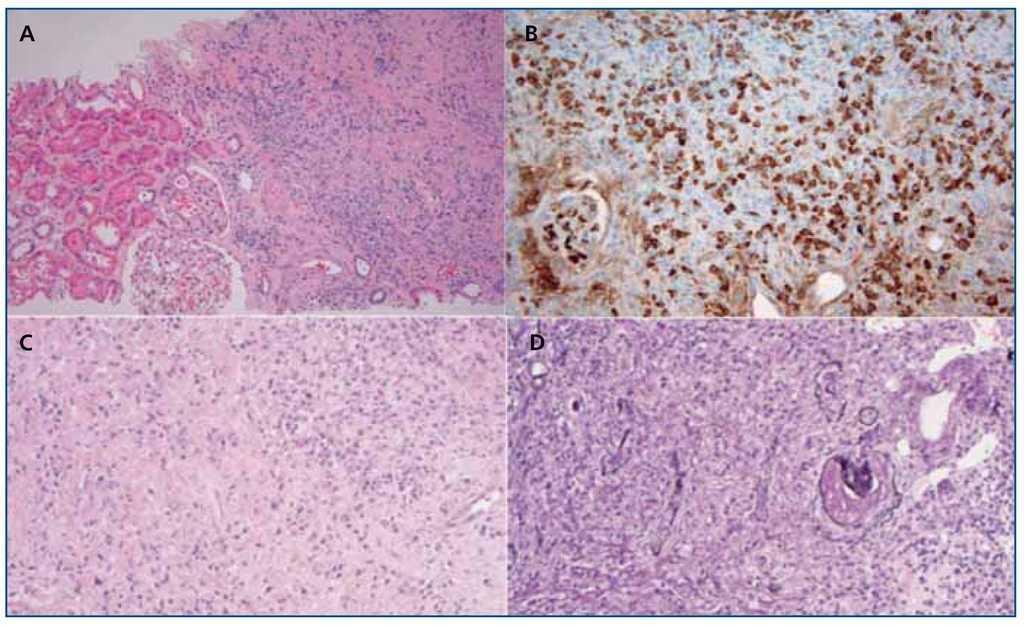
Figura 3. A) Lesiones tubulointersticiales bien diferenciadas del tejido sano adyacente; solo unas pocas células inflamatorias se extienden al parénquima sano (hematoxilina-eosina); B) Inmunohistoquímica: tinción IgG4 de una NTI-IgG4: denso infiltrado uniforme de células plasmáticas IgG4+. Adaptado con permiso de: Macmillan Publishers Ltd: Modern Pathology45, copyright 2011; C) Infiltrado inflamatorio rico en células plasmáticas con numerosos eosinófilos y un patrón de fibrosis estoriforme (hematoxilina-eosina); D) Membranas basales tubulares residuales en medio de un proceso fibroinflamatorio intersticial destructivo (plata metenamina-Jones). Adaptado con permiso de: Macmillan Publishers Ltd: Kidney Int30, copyright 2013.
La fibrosis «estoriforme» es una característica histopatológica esencial y se describe como un patrón irregular de fibrosis que se asemeja a los radios de la rueda de un carro, con células fusiformes que irradian desde el centro10,19,20,22,23 (figura 3 C).
Se ve todo un espectro histolopatológico que va desde un denso infiltrado celular con poca fibrosis hasta un patrón paucicelular densamente fibrótico y expansible que empuja y destruye los túbulos, atrofiándolos20, por lo que en algunos casos solo se aprecian fragmentos de membranas basales tubulares23 (figura 3 D).
Algunos pacientes presentan una NTI aguda con mínima fibrosis en la que se aprecian unos nidos de células inflamatorias con fibras irregulares alrededor que recuerdan el ojo de un pájaro o la veta de la madera de arce. Es una lesión desconocida en tejidos extrarrenales, ya que ha sido descrita con las tinciones de plata metenamina y ácido periódico de Schiff (PAS)20,47.
La infiltración por eosinófilos, la distribución regional de las lesiones (figura 3 A) y la extensión de estas a la cápsula renal completan los datos patológicos22,47. A veces, el proceso esclerosante provoca la dilatación quística de los túbulos42.
La flebitis obliterante, característica fundamental de las IgG4-RD, rara vez se observa en las NTI-IgG4, probablemente debido al pequeño tamaño de las muestras obtenidas por biopsia con aguja del riñón47.
Microscopio de inmunofluorescencia (IF): detecta depósitos de inmunocomplejos en la membrana basal tubular (MBT) en el 50-80 % de los pacientes. Son depósitos granulares, focales o difusos, de IgG (e IgG4 con inmunoperoxidasa) (figura 3 B), cadenas ligeras kappa y lambda y/o C3 de menor intensidad, y ocasionalmente C1q, que se localizan en las zonas de fibrosis intersticial y no aparecen en las áreas sanas20,47.
Microscopio electrónico (ME): detecta depósitos amorfos electrón-densos en la MBT en > 80 % de los pacientes que corresponden principalmente a IgG4 y en menor medida a IgG1 e IgG320,47. No hay depósitos glomerulares a menos que se asocie con una glomerulonefritis por inmunocomplejos23.
Lesiones glomerulares (NM-IgG4)
MO: el glomérulo muestra asas capilares engrosadas. Con tinciones de PAS o plata se ponen de relieve los spikes en la membrana basal glomerular y con el tricrómico de Mason se ven depósitos subepiteliales (inmunocomplejos) e incluso se ha descrito un patrón segmentario de hipercelularidad endocapilar9,30,31 (figura 4 A).
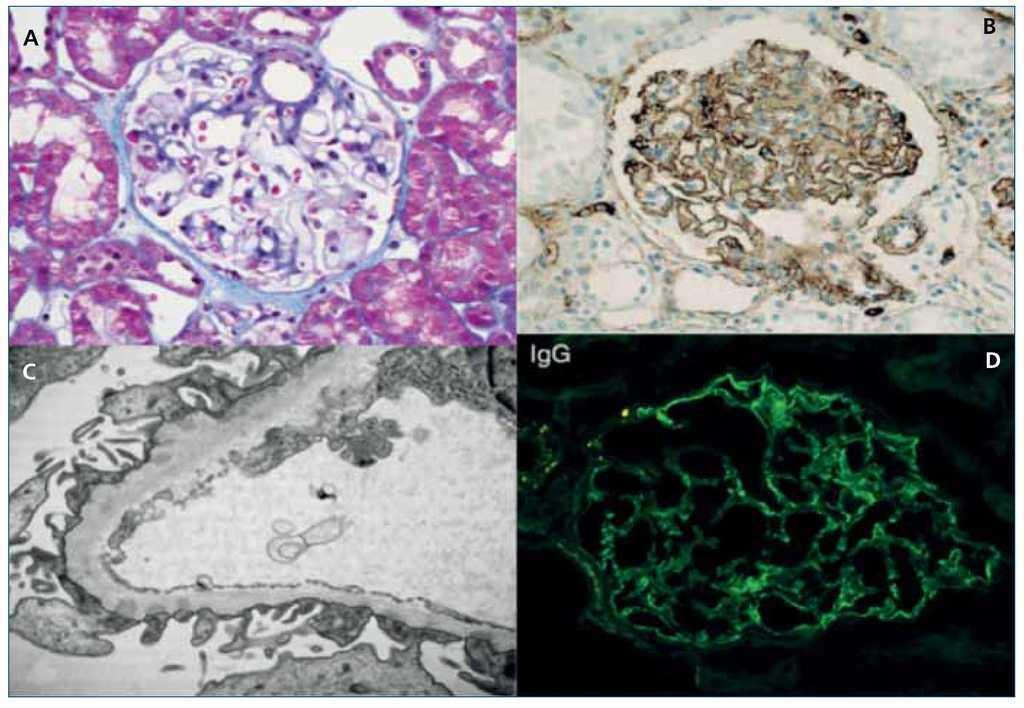
Figura 4. A) Glomérulo aparentemente normal (tricrómico de Masson); B) Depósitos de células plasmáticas IgG4+ en el glomérulo (inmunohistoquímica). Adaptado con permiso de: Macmillan Publishers Ltd: Kidney Int19, copyright 2010; C) Depósitos subepiteliales en la membrana basal glomerular (microscopio electrónico); D) Depósitos granulares de IgG (inmunofluorescencia). Adaptado con permiso de: Macmillan Publishers Ltd: Kidney Int30, copyright 2013.
IF: los glomérulos muestran típicamente depósitos granulares segmentarios o globales para IgG (figura 4 D), C3 y cadenas kappa y lambda y, si se utilizan inmunotinciones para subclases de IgG o inmunohistoquímica, se ven los depósitos glomerulares de IgG4 (figura 4 B). La inmunotinción para PLA2R es negativa. Se estima que un 56 % tienen además datos de NTI-IgG4 y, comparando con la NTI-IgG4, los depósitos de inmunocomplejos en la MBT son menos frecuentes en los casos asociados con NM y están presentes solo en el 33 % de los casos23.
ME: depósitos subepiteliales electrón-densos en la membrana basal glomerular23 (figura 4 C).
Lesiones vasculares
Recientemente se ha publicado una arteritis renal de células plasmáticas en un paciente con NTI-IgG4. Esta lesión afecta a arterias de pequeño y mediano tamaño, y provoca una inflamación marcada de la íntima, media y adventicia por células plasmáticas y linfocitos. Muchas células plasmáticas IgG4 están presentes en la pared arterial y no se observa necrosis fibrinoide ni ruptura de la capa elástica ni neutrófilos28.
CRITERIOS DIAGNÓSTICOS
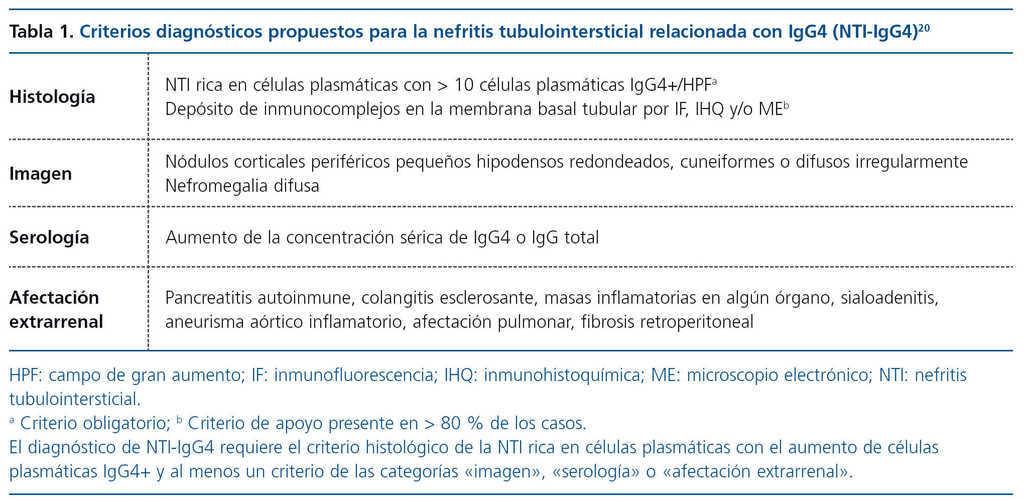
Se exponen los de un grupo multicéntrico norteamericano (tabla 1) y el de la Sociedad Japonesa de Nefrología (tabla 2).

DIAGNÓSTICO DIFERENCIAL
Se apoya fundamentalmente en los hallazgos histopatológicos, aunque deben contemplarse también las características clínicas y radiológicas para diferenciarlos de otros trastornos autoinmunes, inflamatorios o cáncer. Así, enfermedades sistémicas como el lupus eritematoso sistémico, el síndrome de Sjögren, la crioglobulinemia o la sarcoidosis pueden plantear dudas diagnósticas por su similitud clínica que se resuelven con el nivel de IgG4 en suero o en el tejido20-22,25. Otros procesos como la granulomatosis con poliangeítis, la nefropatía diabética, la NTI idiopática, la NM idiopática o incluso la nefritis lúpica pueden compartir niveles séricos elevados de IgG4 que no son exclusivos de las IgG4-RD1,45. Desde el punto de vista radiológico, se deben excluir linfomas, la enfermedad de Castleman multicéntrica, el carcinoma del tracto urinario o incluso pielonefritis.
TRATAMIENTO
Los corticosteroides son el tratamiento de primera línea, mejoran los síntomas, reducen las megalias, optimizan la función del órgano y disminuyen los niveles séricos de IgG4 en pocas semanas. La rápida respuesta al tratamiento con esteroides es un rasgo característico de las IgG4-RD25.
Se han visto efectos positivos de los esteroides sobre la fibrosis activa no avanzada, al reducir el factor de crecimiento del tejido conectivo (particularmente abundante en las zonas de lesión) y actuar sobre las células Treg48.
En casos de corticorresistencia o coticodependencia puede utilizarse rituximab21. En las dosis habituales logra reducir las concentraciones séricas de IgG4 preservando relativamente el nivel de las otras subclases de Ig, lo que sugiere que depleciona las células plasmáticas de vida media corta que están secretando IgG4 y una vez que estas desaparecen del plasma no se replecionan debido al efecto anti-CD20 que agota el pool de células B circulantes49.
Otras opciones, generalmente asociadas a esteroides, son azatioprina (1,5-2 mg/kg/día) o micofenolato mofetil (hasta 2,5 g/día)1.
Los corticosteroides son eficaces para la mayoría de los casos19,20,23-25 incluso si hay fibrosis intersticial grave20,23. En dosis de 0,5-0,75 mg/kg/día en inducción durante 2-4 semanas mejora el filtrado glomerular (FG), los niveles de complemento y los hallazgos radiológicos renales en un mes en la mayoría de los pacientes19. Tras una pauta descendente y un mantenimiento en dosis baja durante meses e incluso años, el FG puede permanecer estable durante bastante tiempo25.
La NM-IgG4 no tiene un tratamiento establecido. Como en otras nefropatías proteinúricas, se deben pautar inhibidores del sistema renina angiotensina aldosterona. Se han descrito remisiones, iniciales o tardías, con esteroides, solos o en combinación con micofenolato mofetil o ciclofosfamida. También se han utilizado rituximab y ciclosporina30.
En algunos casos puede ser necesaria la cirugía41.
EVOLUCIÓN Y PRONÓSTICO
La remisión espontánea es rara y las recaídas son frecuentes (20-30 %) al reducir la dosis o suspender el tratamiento. Pueden detectarse precozmente ante la persistencia de niveles séricos elevados de IgG4 o la reaparición de hipocomplementemia tras el tratamiento con esteroides25.
A veces no se obtiene la respuesta esperada, quizá en casos con fibrosis avanzada1,27. Tras una pauta descendente y un mantenimiento en dosis baja durante meses e incluso años, el FG puede permanecer estable durante bastante tiempo.
Parece haber mayor riesgo de cáncer (sobre todo, gástricos, también pulmón, próstata, colon, linfoma no Hodgkin, conducto biliar y tiroides) en las pancreatitis relacionadas con IgG4, especialmente en el año tras el diagnóstico, lo que apuntaría a que la IgG4-RD sería síndrome paraneoplásico en algunos pacientes50.
CRITERIOS CON LOS QUE SE HA REALIZADO LA REVISIÓN
Se han buscado en pubMed artículos con los términos «IgG4, IgG4 related disease, IgG4 related kidney disease, tubulointerstitial nephritis, membranous nephropathy», publicados en inglés y sin límite de tiempo.
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.
Correspondencia:
José M. Baltar Martín
Sección de Nefrología.
Hospital de San Agustín.
Camino de Los Heros, 6. 33401, Avilés, Asturias.
jbaltarm@telecable.es


