Activation of the focal adhesion kinase (FAK) in podocytes is involved in the pathogenesis of minimal change disease (MCD), but the pathway leading to its activation in this disease is unknown. Here, we tested whether podocyte β1 integrin is the upstream modulator of FAK activation and podocyte injury in experimental models of MCD-like injury.
MethodsWe used lipopolysaccharide (LPS) and MCD sera to induce MCD-like changes in vivo and in cultured human podocytes, respectively. We performed functional studies using specific β1 integrin inhibitors in vivo and in vitro, and integrated histological analysis, western blotting, and immunofluorescence to assess for morphological and molecular changes in podocytes. By ELISA, we measured serum LPS levels in 35 children with MCD or presumed MCD (idiopathic nephrotic syndrome [INS]) and in 18 healthy controls.
ResultsLPS-injected mice showed morphological (foot process effacement, and normal appearing glomeruli on light microscopy) and molecular features (synaptopodin loss, nephrin mislocalization, FAK phosphorylation) characteristic of human MCD. Administration of a β1 integrin inhibitor to mice abrogated FAK phosphorylation, and ameliorated proteinuria and podocyte injury following LPS. Children with MCD/INS in relapse had higher serum LPS levels than controls. In cultured human podocytes, β1 integrin blockade prevented cytoskeletal rearrangements following exposure to MCD sera in relapse.
ConclusionsPodocyte β1 integrin activation is an upstream mediator of FAK phosphorylation and podocyte injury in models of MCD-like injury.
La activación de la quinasa de adhesión focal (FAK) en podocitos juega un papel en la patogénesis de la enfermedad de cambios mínimos (ECM), pero su mecanismo de activación en dicha enfermedad es desconocido. En este estudio investigamos si la integrina β1 de los podocitos modula la activación de FAK y del daño podocitario en modelos experimentales de la ECM.
MétodosUtilizamos lipopolisacárido (LPS) y suero de pacientes con ECM para inducir daño podocitario in vivo e in vitro, respectivamente. Realizamos estudios funcionales usando inhibidores específicos de la integrina β1 in vivo e in vitro, así como estudios histológicos, western blots y técnicas de inmunofluorescencia para evaluar cambios morfológicos y moleculares en podocitos. Usando ELISA medimos los niveles séricos de LPS en 35 niños con ECM o sospecha de ECM (síndrome nefrótico idiopático [SNI]) y en 18 individuos sanos.
ResultadosLos ratones inyectados con LPS desarrollaron cambios morfológicos (fusión de pedicelos, con apariencia normal de los glomérulos) y moleculares (pérdida de la expresión de sinaptopodina, cambio en la localización de la nefrina fosforilada y fosforilzación de FAK), que son característicos de la ECM en humanos. La administración de un inhibidor de la integrina β1 en ratones disminuyó la fosforilación de FAK, proteinuria y daño podocitario que ocurre tras la inyección de LPS. En niños con ECM/SNI, los niveles séricos de LPS fueron más elevados que en controles. En cultivos de podocitos humanos, la adicción de un inhibidor de la integrina β1 al suero de niños con ECM en recaída evitó cambios en el citoesqueleto.
ConclusionesLa integrina β1 de los podocitos actúa como mediador de la activación de la FAK y del daño podocitario en modelos experimentales de la ECM.
Minimal change disease (MCD) is the most common type of nephrotic syndrome in children and is thought to be mediated by circulating factors that directly target podocytes.1,2 Several candidates have been postulated,3–5 but the signaling pathways initiating podocyte injury in MCD remain poorly understood.
Upon injury, podocytes usually exhibit retraction of their foot processes, known as foot process effacement (FPE), as well as molecular alterations.6 Specifically, MCD is associated with extensive FPE, synaptopodin loss, nephrin de-phosphorylation, nephrin loss and/or mislocalization, and phosphorylation of the focal adhesion kinase (FAK).7–10
Podocyte FAK mediates proteinuria and FPE in several experimental models of podocyte injury.11,12 FAK can be activated by multiple mechanisms.13 In experimental models, nephrin phosphorylation mediates podocyte FAK activation,14 but the observation that nephrin phosphorylation is reduced in MCD10 suggests that other signaling pathways are involved in podocyte FAK activation in MCD.
In this study, we combined animal and cell culture studies to test the hypothesis that β1 integrin is the upstream signal for podocyte FAK activation and podocyte injury in experimental models of MCD-like injury.
Materials and methodsReagents and antibodiesThe following primary antibodies were used: monoclonal mouse anti-synaptopodin (10R-2373, Fitzgerald) at 1:20 dilution; purified polyclonal rabbit anti-nephrin (kindly provided by Rakesh Verma) at 1:100; monoclonal rabbit anti-nephrin phospho Y1176+Y1193 (ab80299, Abcam) at 1:100; polyclonal rabbit anti-podocin (P0372, Sigma–Aldrich) at 1:100; monoclonal rat anti-mouse CD29-activated β1 integrin – (550531, clone 9EG7, BD Pharmigen) at 1:100; monoclonal rabbit anti-phospho-FAK (Tyr397) (31H5L17, ThermoFisher) at 1:500; monoclonal rabbit anti-GAPDH (2118, cell signaling) at 1:7000. The following reagents were used: lipopolysaccharide (LPS) from Escherichia Coli O111:B4 (Sigma–Aldrich); monoclonal hamster anti CD29-β1 integrin inhibitor for animal studies – (HMβ1, Biolegend),15 and Armenian hamster Ig G negative control (MCA2356, Biorad) used for intraperitoneal (IP) injections; monoclonal hamster anti CD29 (HMβ1, Biolegend) conjugated with 10-nm gold particles (approximately 4×1013particles/ml, Aurion). For in vitro assays, we used a purified rat anti-human CD29 (Mab13, 552828, BD Pharmigen); fibronectin (356008, BD Pharmigen); and Rhodamine Phalloidin (R415, ThermoFisher).
LPS mouse model and β1 integrin blockade in vivoEight- to twelve-week-old B6 background mice were injected with LPS (10μg/g body weight in 200μl phosphate buffered saline [PBS]) or an equal volume of sterile PBS intraperitoneally. Spot urine samples were collected before and after LPS injection at different time points. Albuminuria was measured using ELISA and a polyclonal goat anti-mouse albumin (A90-134A, Bethyl laboratories), and results were normalized to urine creatinine. A subset of mice was injected intraperitoneally with a monoclonal hamster antibody (allosteric inhibitor) against mouse β1 integrin (HMβ1, Biolegend, 2.5μg/g)15 or Armenian hamster IgG (control) 20hours prior to LPS injection, as described above. In these mice, albuminuria was also measured prior to and 20h following HMβ1 injection, but prior to LPS injection.
ImmunostainingFresh frozen mouse kidneys were sectioned at 5-μm, and acetone fixed for eight minutes. Sections were blocked with 10% goat serum for 1h. Frozen slides were used for immunofluorescence of mouse kidney tissue with activated β1 integrin (9EG7) and podocin. For paraffin embedded mouse kidney, tissue samples were fixed in 10% formalin, paraffin embedded, sectioned at 3-μm, deparaffinized and rehydrated. Antigen retrieval was performed with 1X Tris–EDTA buffer at 96°C for 30min. After 20-min time to cool down, residual aldehydes were quenched in 0.1% sodium borohydride in Sorenson buffer for 15min and permeabilized in 0.1% Triton X-100 for 10min. After three 1× PBS washes, slides were blocked with 1% Bovine Serum Albumin (BSA) for 1h. This was followed by overnight incubation at 4°C with the corresponding primary antibody. For frozen and paraffin embedded slides, overnight incubation with primary antibody was followed by three 1× PBS washes and incubation with the appropriate Alexa Fluor secondary antibodies at 1:400 for 2h at room temperature. Slides were washed again with 1× PBS and mounted with Vectashield® Mounting Medium with DAPI4,6,-Diamidino-2-phenylindole (DAPI). Images were captured with confocal microscopy for frozen tissue using a Leica Inverted SP53 Confocal Microscope and using a non-confocal Leica Microscope for paraffin embedded tissue.
Transmission electron microscopy (TEM)Mouse kidney tissue preparation and processing was done using standard techniques.
Immunogold electron microscopyThe hamster monoclonal antibody against mouse β1 integrin (HMβ1) was conjugated to 10nm-gold particles by Aurion (https://aurion.nl/). Tail vein injection of this conjugated primary antibody at 2.5μg/g body weight was performed and kidneys were harvested 20min later. Tissue was fixed with 2% paraformaldehyde and 2.5% glutaraldehyde. Samples were processed and analyzed using a JEOL JEM-1400Plus Transmission Electron Microscope.
Western blot analysisProtein concentrations from isolated mouse glomeruli were determined using a BCA protein assay (Pierce). Proteins were extracted from plasma membranes in RIPA buffer (PBS containing 0.1% SDS, 1% Nonidet P-40, 0.5% sodium deoxycholate and 100mM potassium iodide). Lysates were resolved using SDS-PAGE and transferred to PVDF membrane (GE Healthcare) using semidry transfer (Bio-Rad). Membranes were blocked using 5% milk. Immunoblotting was performed with the indicated primary (phospho FAK and GAPDH) antibody followed by secondary antibody.
Cell cultureConditionally immortalized human podocytes were a gift from Dr. Saleem (University of Bristol, United Kingdom). Podocytes were cultured in RPMI medium with Glutamax (Invitrogen), 10% fetal bovine serum (FBS) (Invitrogen Corp), 200U/ml penicillin, 0.1mg/ml streptomycin (Roche Applied Science) along with ITS (insulin, transferrin, and selenium) (Invitrogen Corp) per standard protocols. For experiments, six well culture plates were coated with fibronectin (1mg/ml diluted in PBS) and incubated at 37°C for 2h. Plates were then washed with PBS prior to plating podocytes in complete media.
Human podocytes stimulation and β1 integrin blocking assaysDifferentiated human podocytes were cultured with 20% sera from children with MCD or idiopathic nephrotic syndrome (INS), in relapse and remission, for 24h. Subsequently, plates were washed with PBS and fixed with 4% paraformaldehyde. Next, cells were permeabilized using 0.5% Triton X-100 in PBS at room temperature for 5min followed by PBS washes and blocking step with 1% BSA for 1h. Next, we used Rhodamine Phalloidin (1:100 dilution) to assess changes in fine F-actin stress fibers. For blocking assays, podocytes were placed on fibronectin coated plates as above. Prior to MCD/INS sera treatments, a β1 integrin inhibitor (Mab13 antibody)16 was mixed with culture media at 0.1ml/ml concentration for 20min at room temperature. This was followed by PBS washes, MCD/INS sera treatments, and phalloidin staining as above.
Human studiesDefinitions. We used standard definitions for MCD, INS, relapse and remission.17Participants. Demographics and clinical data are shown in Supplementary Table 1. There were 35 children with diagnosis of MCD or INS at time of disease onset or during relapse. We included INS because kidney biopsies are not routinely performed in these children, but it is usually associated with MCD.18 Eighteen children without known history of glomerular disease or proteinuria served as control subjects. Patients and control subjects were recruited at 5 institutions (study approvals shown in ‘Statement of Ethics’). Five control serum samples were obtained from Precision for Medicine (precisionformedicine.org). LPS measurement. Blood samples were collected, processed and stored at −80°C. All samples were serum except for 5 plasma samples in the control group. LPS quantification was performed using an ELISA kit (CSB-E09945h, Cusabio) as per manufacturer recommendations.
Statement of ethicsHuman studies: The study was was conducted ethically in accordance with the World Medical Association Declaration of Helsinki and was approved by the Colorado Multiple Institutional Review Board (#13-2700) at the University of Colorado and collaborative institutions: Rocky Mountain Kidney Center (#13-2700), Hospital Universitario Central de Asturias (#221/19), Hospital Niño Jesus (#R-0011/20), and Hospital Universitario Santa Lucia (EO-39/21). Written informed consents, and assents if appropriate, were obtained from participants and parents/guardian. Animal studies: All animal studies were approved by the Institutional Animal Care and Use Committee at the University of Colorado, approval number #1038, and experiments were carried out in accordance with the National Insitutes of Health guide for the care and use of Laboratory animals.
Statistical analysisStatistical analysis was performed using GraphPad Prism (version 9, GraphPad Software). Due to non-normal distribution, so we examined differences among two groups using the unpaired two-tailed Mann–Whitney U. Mouse proteinuria was expressed as mean±standard error of the mean (s.e.m). LPS concentration in human sera was expressed as median±interquartile range. ImageJ software was used to quantify the fluorescence in ≥10 glomeruli per animal in mouse studies. For quantification of stress fibers in cultured human podocytes, we evaluated cells in ten fields at ×20 magnification, and stress fibers were expressed as percentage of cells with loss of actin filaments across the cytoplasm. An alpha<0.05 was considered statistically significant.
ResultsLPS replicates some key features of human MCDTo test our hypothesis, we first evaluated if the LPS model of podocyte injury replicates some key features of human MCD. Among the two most common animal models to study MCD, LPS and puromycin aminoglycoside (PAN),19 we chose the former because infections usually trigger MCD relapse in children,20 and because PAN is associated with glomerulosclerosis.21 Compared to controls (PBS), LPS-injected mice developed transient albuminuria (shown in Fig. 1a) and foot process effacement with normal appearing glomeruli on light microscopy (shown in Fig. 1b). By immunofluorescence, synaptopodin was decreased in LPS mice compared to controls, whereas the expression of total nephrin was preserved during injury (shown in Fig. 1c). Next, we performed dual immunofluorescence for phosphorylated nephrin and synaptopodin to assess for changes in the phosphorylation status and localization of nephrin during injury and recovery. At the peak of proteinuria, nephrin phosphorylation was increased (shown in Fig. 1d), compared to controls, but also mislocalized as noted by its granular pattern (shown in Fig. 1d, far right column, arrows). Notably, nephrin exhibited a peak of phosphorylation and recovered its lineal pattern coinciding with the recovery phase of proteinuria (shown in Fig. 1d, far right column, arrowheads).
 LPS causes transient albuminuria reaching a peak at 24h. N=4–6 per group. * p=0.004; ** p=0.002; *** p=0.009, compared to control group (time 0). Data presented as mean±s.e.m. (b) By TEM (upper lane), LPS induces partial foot processes effacement by 24h. Original images captured at 20,000×. Scale bars: 100μm. Middle lane shows a magnification of the upper lane images marked with a white square. By PAS staining (lower lane), mouse glomeruli appear normal or near normal following LPS. N=3–5 per group. Images captured at 40×. Scale bars: 500μm. TEM transmission electron microscopy; PAS Periodic acid-Schiff; WT wild-type mice (controls). (c) LPS is associated with synaptopodin loss, whereas the expression of total nephrin remains intact following injury. N=3–5 per group. *p=0.02, ns not statistically significant. Images captured at 60×. Scale bars: 50μm. (d) LPS increases nephrin phosphorylation over time (p<0.01 for time 0 vs 24h and 0 vs 36h). Far right column shows mislocalization of phosphorylated nephrin as noted by its granular pattern (arrows) 24h after LPS, and a normal lineal pattern (arrowheads) prior to that and during the resolution phase of proteinuria. * p=0.02. N=3–5 per group. Images captured at 60×. Scale bars: 50μm. (e) LPS levels are higher in serum from children with MCD/INS during relapse (N=35) than in control subjects (N=18). * p=0.01. Data presented as median±interquartile. MCD, minimal change disease; INS, idiopathic nephrotic syndrome. Red dots represent patients with new onset INS.")
LPS replicates some key features of human MCD. (a) LPS causes transient albuminuria reaching a peak at 24h. N=4–6 per group. * p=0.004; ** p=0.002; *** p=0.009, compared to control group (time 0). Data presented as mean±s.e.m. (b) By TEM (upper lane), LPS induces partial foot processes effacement by 24h. Original images captured at 20,000×. Scale bars: 100μm. Middle lane shows a magnification of the upper lane images marked with a white square. By PAS staining (lower lane), mouse glomeruli appear normal or near normal following LPS. N=3–5 per group. Images captured at 40×. Scale bars: 500μm. TEM transmission electron microscopy; PAS Periodic acid-Schiff; WT wild-type mice (controls). (c) LPS is associated with synaptopodin loss, whereas the expression of total nephrin remains intact following injury. N=3–5 per group. *p=0.02, ns not statistically significant. Images captured at 60×. Scale bars: 50μm. (d) LPS increases nephrin phosphorylation over time (p<0.01 for time 0 vs 24h and 0 vs 36h). Far right column shows mislocalization of phosphorylated nephrin as noted by its granular pattern (arrows) 24h after LPS, and a normal lineal pattern (arrowheads) prior to that and during the resolution phase of proteinuria. * p=0.02. N=3–5 per group. Images captured at 60×. Scale bars: 50μm. (e) LPS levels are higher in serum from children with MCD/INS during relapse (N=35) than in control subjects (N=18). * p=0.01. Data presented as median±interquartile. MCD, minimal change disease; INS, idiopathic nephrotic syndrome. Red dots represent patients with new onset INS.
To further assess the potential relevance of the model to human MCD, we next measured LPS levels, using ELISA, in serum from children with MCD and clinically resumed MCD (INS) during relapse and in healthy controls. We found that serum LPS was significantly higher in MCD/INScompared to controls (shown in Fig. 1e).
Podocyte β1 integrin signaling mediates FAK activation and podocyte injury in vivoWe first examined if podocyte β1 integrin and FAK are activated following LPS. To assess this, we performed immunostaining of mouse kidney tissue using an antibody against activated β1 integrin and podocin, as a podocyte marker, as well as western blotting of glomerular lysates testing for phosphorylated FAK. Following LPS, we found an increased in activated β1 integrin expression in podocytes, as noted by its colocalization with podocin (shown in Fig. 2a), and an increase in FAK phosphorylation (shown in Fig. 2b). We next asked whether β1 integrin activation could be an important upstream mediator of FAK phosphorylation and podocyte injury in this model. To address this, we administered a well-characterized specific blocking antibody to β1 integrin, known as HMβ1,15 into LPS and control mice. We initially tested the antibody in wild-type mice, and we found that it was well tolerated and bound specifically to the major types of glomerular cells in the kidney (endothelial, podocyte, mesangial cells), but not to tubular cells (shown in Fig. 3a, left upper corner). Specific binding to podocytes was shown using both immunoelectron microscopy involving HMβ1-conjugated with 10nM gold particles (shown in Fig. 3a, upper row) and double immunostaining with podocin, as podocyte marker (shown in Fig. 3a, bottom row). When injected alone, HMβ1 did not cause changes in baseline proteinuria in healthy mice (shown in Supplementary Fig. 1). Next, we pre-treated mice with a single intraperitoneal dose of HMβ1 or IgG isotype (control) followed by a single dose of LPS 20h later. The administration of HMβ1 ameliorated proteinuria (shown in Fig. 3b), prevented FAK phosphorylation (shown in Fig. 3c), and preserved podocyte health and shape, as noted by the preserved synaptopodin expression and podocyte foot process morphology, respectively, compared to control group (shown in Fig. 3d, upper and lower row respectively).
 Frozen kidney sections from control and LPS-treated mice were stained using an antibody against activated β1 integrin. Podocin was used a podocyte marker. Activated β1 integrin expression colocalized with podocin 24h following LPS, but not at baseline. N=3–5 per group. Imaged were captured at 60×. Scale bars: 100μm. (b) By western blot of glomerular lysates, phosphorylated FAK was upregulated 24h following LPS. N=3 per group. WT, wild-type mice (controls).")
LPS activates podocyte β1 integrin and FAK in vivo. (a) Frozen kidney sections from control and LPS-treated mice were stained using an antibody against activated β1 integrin. Podocin was used a podocyte marker. Activated β1 integrin expression colocalized with podocin 24h following LPS, but not at baseline. N=3–5 per group. Imaged were captured at 60×. Scale bars: 100μm. (b) By western blot of glomerular lysates, phosphorylated FAK was upregulated 24h following LPS. N=3 per group. WT, wild-type mice (controls).
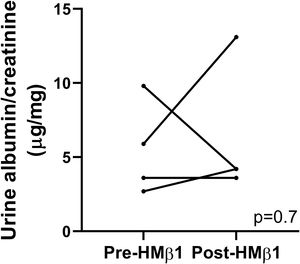
 HMβ1 (green) bound glomerular but not tubular cells by immunofluorescence of mouse kidney (upper row, left side). Images captured at 20×. Scale bars: 100μm. By TEM, injected HMβ1 conjugated with 10-nm gold particles (arrowheads) deposited onto glomerular endothelial cells and to the basal aspect of podocytes (upper row). Original images captured at 20,000×. N=1. HMβ1 bound podocytes as shown by double immunostaining using podocin as podocyte marker (lower row). (b) Pre-treatment with the β1 integrin inhibitor HMβ1 ameliorated LPS-induced albuminuria. N=8 per group. *p=0.01. (c) HMβ1 prevented FAK phosphorylation in glomerular lysate following LPS. N=3–5 per group. This blot was cropped from the same gel displayed in Fig. 2b. (d) HMβ1 preserved synaptopodin expression (*p=0.02) and podocyte foot processes morphology, by transmission electron microscopy, in mice pre-treated with HMβ1 compared to Ig G controls, following LPS. N=3–5 per group. Bottom lane shows a magnification of the area marked with a black square in the middle lane. Image captured at 60×. Scale bars: 500μm.")
Podocyte β1 integrin is an upstream mediator of FAK activation and podocyte injury in vivo. (a) HMβ1 (green) bound glomerular but not tubular cells by immunofluorescence of mouse kidney (upper row, left side). Images captured at 20×. Scale bars: 100μm. By TEM, injected HMβ1 conjugated with 10-nm gold particles (arrowheads) deposited onto glomerular endothelial cells and to the basal aspect of podocytes (upper row). Original images captured at 20,000×. N=1. HMβ1 bound podocytes as shown by double immunostaining using podocin as podocyte marker (lower row). (b) Pre-treatment with the β1 integrin inhibitor HMβ1 ameliorated LPS-induced albuminuria. N=8 per group. *p=0.01. (c) HMβ1 prevented FAK phosphorylation in glomerular lysate following LPS. N=3–5 per group. This blot was cropped from the same gel displayed in Fig. 2b. (d) HMβ1 preserved synaptopodin expression (*p=0.02) and podocyte foot processes morphology, by transmission electron microscopy, in mice pre-treated with HMβ1 compared to Ig G controls, following LPS. N=3–5 per group. Bottom lane shows a magnification of the area marked with a black square in the middle lane. Image captured at 60×. Scale bars: 500μm.
MCD sera in relapse has been previously shown to activate podocytes,22,23 consistent with the idea that the sera may carry circulating pathogenic factors. To test whether the podocyte β1 integrin is a target of those circulating factors and mediator of podocyte injury, we performed functional studies using cultured human podocytes, MCD sera in relapse and remission, and the human β1 integrin-blocking antibody Mab13.16 Sera from children with MCD in relapse, but not in remission, caused actin rearrangement in cultured human podocytes, and this was abrogated by preemptive incubation with Mab13 (shown in Fig. 4).
 leads to actin rearrangement (arrowheads) compared to sera in remission (RM). N=4 per group. *p=0.02. Mab13 prevents changes in actin stress fibers (white arrows) in podocytes cultured with MCD sera in relapse. N=3. **p=0.04. Scale bars: 100μm. Data presented as mean±S.D. MCD, minimal change disease.")
MCD sera in relapse mediate podocyte injury via β1 integrin signaling. Sera from children with MCD in relapse (RL) leads to actin rearrangement (arrowheads) compared to sera in remission (RM). N=4 per group. *p=0.02. Mab13 prevents changes in actin stress fibers (white arrows) in podocytes cultured with MCD sera in relapse. N=3. **p=0.04. Scale bars: 100μm. Data presented as mean±S.D. MCD, minimal change disease.
Minimal change disease is considered a podocyte disease mediated by circulating factors, yet the mechanisms initiating podocyte injury remain incompletely understood.1 By integrating animal and cell culture studies, here we showed that podocyte β1 integrin signaling mediates podocyte injury in a traditional animal model of MCD-like injury, that replicates some key features of human MCD, and in cultured human podocytes.
In this study, we demonstrated that podocyte β1 integrin activation and downstream signaling involving FAK phosphorylation is an important causal pathway for podocyte injury in vivo. We showed that the β1 integrin inhibitor HMβ115 ameliorated podocyte injury, as noted by the reduction in proteinuria, abrogation of FAK phosphorylation, and preservation of synaptopodin expression and podocyte foot processes. Our data identify podocyte β1 integrin signaling as upstream mediator of podocyte injury in this model of proteinuria, raising the possibility that circulating factors involved in MCD may cause podocyte injury by activating this pathway. Consistent with this, we showed that MCD sera in relapse can directly cause podocyte injury in vitro, as noted by actin rearrangements, and this was abrogated by β1 integrin blockade. Collectively, our data support the hypothesis that podocyte β1 integrins may be a molecular target for circulating factors in MCD, and this may help guide the search for novel circulating factors important for the disease.
Contrary to our findings, Lee et al. reported a reduction in activated β1 integrin expression in LPS mice, and that a β1 integrin agonist ameliorated podocyte injury in vivo.24 These contrasting results may be due to different methodology or off-target effects of the antibody. Notably, we showed that the β1 integrin inhibitor HMβ1 bound specifically to podocytes and prevented morphological and molecular alterations in vivo, thus supporting a protective role directly on podocytes. In support to this, injection of the β1 integrin inhibitor HMβ1 into mice does not ameliorate the systemic immune response following LPS.15 Consistent with our findings, Ma et al. also showed that LPS triggers podocyte FAK phosphorylation in mice.11 Genetic and pharmacological inhibition of α2β1 integrins also ameliorated other forms of glomerular injury in vivo.25 Thus, it is possible that the effects of β1 integrin inhibitors or agonists may depend on the targeted cell and/or the specific α chain associated to β1 integrin. In humans, podocyte β1 integrin upregulation has been reported in early diabetic nephropathy,26 whereas loss of podocyte β1 integrin activation is observed in focal segmental glomerulosclerosis (FSGS), but not in MCD.27 Consistent with this, podocyte FAK phosphorylation is increased in MCD, but not FSGS.9
Our study is the first to show that LPS levels are high in serum from children with MCD/SSNS during relapse. This finding seems relevant to MCD because LPS is not only a marker of systemic inflammation but also a trigger of the innate immune response, which is involved in the disease pathogenesis.28 LPS has been also linked to cardiovascular risk and progression to diabetic nephropathy in patients with type 1 diabetes.29,30 Thus, the source or potential role of LPS in MCD warrants further investigations. Given the association of MCD with infections,20 food intolerance and allergies,31,32 it is possible that LPS in circulation is the result of a high permeable gut (‘leaky gut’), or exogeneous sources such as respiratory infections or allergens. Mechanistically, LPS could either activate toll-like receptors on glomerular cells,33 and/or act as amplifier of the immune response or the putative circulating factors.29
Our findings led us to postulate that circulating factors may directly activate podocyte β1 integrins and downstream pathways initiating podocyte shape changes, similar to that reported with other integrins such αvβ3.34 This seems a plausible mechanistic pathway for transient proteinuria in LPS mice and healthy children with transient proteinuria during infections. What's the relevance for MCD? It is tentative to postulate that an exaggerated and/or perpetuated involvement of this pathway may initiate podocyte injury in MCD, and this may be further aggravated by additional insults such as autoantibodies,4,5 local cytokines,35 other circulating factors,2 and/or intrinsic podocyte dysregulation (shown in Fig. 5).36 A candidate target for sustained podocyte injury is nephrin. We showed that LPS induced structural and molecular changes in glomeruli like that observed in MCD except for nephrin phosphorylation, which peaked in the LPS model during the recovery phase of proteinuria whereas it is reduced in MCD during relapse.10 Recently Watts et al. showed evidence for nephrin autoantibodies in 29% patients with MCD,5 and experimental models showed that these autoantibodies as well as impaired mechanisms of nephrin phosphorylation can cause podocyte injury.37,38 Thus, podocyte injury in MCD may be the result of a multi-hit process involving different insults that may explain the heterogeneity of the disease.
 may be necessary to initiate, aggravate and/or perpetuate podocyte injury and proteinuria in MCD. MCD, minimal change disease; FAK, focal adhesion kinase; FAK p, phosphorylated focal adhesion kinase; GBM, glomerular basement membrane. Cartoon created with Biorender.com.")
Proposed role of the podocyte β1 integrin-FAK axis in MCD. Infections are the most common trigger of relapse of nephrotic syndrome in children with SSNS and MCD. Our findings suggest that microbial products such LPS can target podocytes leading to cytoskeletal changes and proteinuria via activation of the β1 integrin/FAK axis. Thus, we postulate that microbial products, along with other molecules with the ability to activate this axis, could be the initial triggers leading to podocyte injury in MCD. Likewise, it is also possible that additional insults (circulating autoantibodies or molecules produced by immune or glomerular cells such endothelial cells) may be necessary to initiate, aggravate and/or perpetuate podocyte injury and proteinuria in MCD. MCD, minimal change disease; FAK, focal adhesion kinase; FAK p, phosphorylated focal adhesion kinase; GBM, glomerular basement membrane. Cartoon created with Biorender.com.
Our study has limitations. We identified podocyte β1 integrin as a candidate target for circulating factors, but the nature of these factors is still under investigation. We showed that β1 integrin antibodies bound and protected podocytes, but we cannot exclude that these antibodies also exert a protective effect on other glomerular cells (such as endothelial cells) that may indirectly help prevent podocyte injury. Because we combined mouse and human studies, we used appropriate β1 integrin blocking antibodies (HMβ1 and Mab13, respectively) for each species. While different antibodies could have a distinct effect on cells, both HMβ1 and Mab13 have been shown to block β1 integrin activation.15,16 Given the disease heterogeneity, our studies do not preclude other pathways to be important in MCD. We used common experimental models of MCD,19,23 but further validation of our studies is necessary whenever better animal models of MCD are available.
In summary, we showed that podocyte β1 integrin blockade ameliorated podocyte injury in vivo and in vitro. The identification of molecules or mechanisms involved in podocyte β1 integrin activation could provide insights into the processes triggering MCD, and may help develop targeted therapies.
Data availability statementAll data analyzed during this study are included in this article and its supplementary material files. Further inquiries can be directed to the corresponding author.
Conflict of interestThe authors declare they have no conflict of interest.