


The term acute tubular necrosis was thought to represent a misnomer derived from morphological studies of human necropsies and necrosis was thought to represent an unregulated passive form of cell death which was not amenable to therapeutic manipulation. Recent advances have improved our understanding of cell death in acute kidney injury. First, apoptosis results in cell loss, but does not trigger an inflammatory response. However, clumsy attempts at interfering with apoptosis (e.g. certain caspase inhibitors) may trigger necrosis and, thus, inflammation-mediated kidney injury. Second, and most revolutionary, the concept of regulated necrosis emerged. Several modalities of regulated necrosis were described, such as necroptosis, ferroptosis, pyroptosis and mitochondria permeability transition regulated necrosis. Similar to apoptosis, regulated necrosis is modulated by specific molecules that behave as therapeutic targets. Contrary to apoptosis, regulated necrosis may be extremely pro-inflammatory and, importantly for kidney transplantation, immunogenic. Furthermore, regulated necrosis may trigger synchronized necrosis, in which all cells within a given tubule die in a synchronized manner. We now review the different modalities of regulated necrosis, the evidence for a role in diverse forms of kidney injury and the new opportunities for therapeutic intervention.
La idea de que el término necrosis tubular aguda supone una denominación inapropiada se deriva de estudios morfológicos de necropsias humanas. La opinión generalizada ha sido que la necrosis representa una forma pasiva de muerte celular no regulada que no es susceptible de manipulación terapéutica. Los recientes avances han mejorado nuestra comprensión de la muerte celular en la lesión renal aguda. En primer lugar, la apoptosis origina una pérdida celular, pero no desencadena una respuesta inflamatoria. Sin embargo, los intentos rudimentarios de interferir en la apoptosis (p. ej., con determinados inhibidores de la caspasa) pueden desencadenar una necrosis y, por lo tanto, una lesión renal mediada por inflamación. En segundo lugar, y lo que es más revolucionario, ha surgido el concepto de necrosis regulada. Se han descrito varias modalidades de necrosis regulada como necroptosis, ferroptosis, piroptosis y necrosis regulada por transición de permeabilidad mitocondrial. De forma análoga a la apoptosis, la necrosis regulada se modula a través de moléculas específicas que actúan como dianas terapéuticas. Al contrario que la apoptosis, la necrosis regulada puede ser extremadamente proinflamatoria y, lo que es importante para el trasplante renal, inmunogénica. Además, la necrosis regulada puede desencadenar una necrosis sincronizada, en la que todas las células del interior de un túbulo concreto mueren de manera sincronizada. Revisaremos las diferentes modalidades de necrosis regulada, la evidencia de una función en las diversas formas de lesión renal y las nuevas oportunidades de intervención terapéutica.
Acute kidney injury (AKI) is characterized by a rapid decline of renal function and its incidence is increasing.1 Causes of AKI include sepsis, ischemia-reperfusion injury, and nephrotoxic agents that induce diverse forms of kidney injury from direct tubular toxicity to crystal-induced kidney injury.2–4 Treatment of AKI is symptomatic and consists of replacement of renal function by dialysis if renal failure is severe. There is not established therapy to accelerate the recovery and attempts at preventing AKI are not universally effective.5 Despite the reversibility, at least partial, of the loss of renal function in most patients that survive, the mortality of AKI remains high (over 50%).6,7 Moreover, AKI episodes favor the progression of chronic kidney disease (CKD)8 and CKD is a risk factor for AKI.9
As CKD, AKI is characterized by loss of renal tubular cells. In fact, it has long been known that tubular cell death is the best histopathological correlate of renal dysfunction in AKI.10,11 In AKI, the initial wave of tubular cell death is followed by compensatory tubular cell proliferation leading to regeneration and by a second wave of cell death that adjusts final cell numbers. Inflammatory cell infiltration and mild fibrosis in a chronic phase are also features of AKI.12 Indeed, dying cells release inflammatory factors that amplify tissue injury. In this regard, targeting inflammatory mediators and receptors protects against ongoing kidney injury in experimental AKI.13–15 Several modalities of cell death have been recently described that are regulated in nature, i.e., requite the activation of specific molecular pathways and can be manipulated therapeutically by targeting these pathways.16 The cell death molecular pathways activated in AKI are still poorly characterized, as is unclear whether they are shared by diverse stimuli or by different time points during the evolution of AKI and CKD.16–18
In the current review, we explore the specific role of different cell death pathways during AKI and their contribution to renal inflammation with emphasis on potential diagnostic and therapeutic implications.
Cell death during AKIFor many years the term acute tubular necrosis was used to refer to AKI characterized by parenchymal renal injury of mainly tubular location. The term acute tubular necrosis was originated in necropsy studies before the phenomena of apoptosis or necrosis had been established as separate forms of cell death. In human acute tubular necrosis, apoptosis was the most common form of tubular cell death from a morphological point of view; albeit necrosis was also present.10,11 We stress the term “morphologically” because some forms of cell death are defined by the response to therapeutic intervention. In this regard, at the time of those studies, regulated necrosis had not been described. Currently, it is well established that several forms of necrosis are regulated processes and different molecular pathways of regulated necrosis have been described. The main difference between apoptosis and necrosis is that apoptosis is not inflammatory because the integrity of the plasma membrane is maintained; while in necrosis plasma membrane integrity is lost leading to the release of molecules that trigger inflammation and immunogenic responses.
Apoptosis was the first type of regulated tubular cell death to be studied and extensively characterized in AKI. Apoptosis can be executed through intrinsic or extrinsic pathways.19 The intrinsic pathway is initiated by cell stress causing outer mitochondrial membrane permeabilization, which results in the release of apoptogenic factors, such as cytochrome c, that then bind Apaf-1 in a multiprotein complex called the apoptosome to activate caspase-9. Bcl-2-related proteins sensitize (e.g. Bax) or protect (e.g. Bcl2, BclxL) from apoptosis. The extrinsic pathway is executed upon ligation of death receptors that recruit adapter proteins and subsequently activate caspase-8.20 Caspase-8 or caspase-9 activation eventually triggers the activation of executioner caspases, such as caspase-3, and cell demolition from the inside. Apoptotic cells express “eat me” signals in the cell surface that are identified by macrophage receptors and receptors expressed in stressed but alive tubular cells, such as KIM-1.21,22 Eventually the apoptotic cell is engulfed by adjacent cells before loss of cell membrane permeability results in release of proinflammatory factors. During kidney injury, apoptosis takes place in different types of intrinsic renal cells including proximal and distal tubular cells, endothelial cells and podocytes.23 Evidence of activation of apoptosis has been observed in different experimental models of AKI and in human AKI, but its contribution to injury is questioned because caspase inhibitors did not offer efficacious protection in most preclinical situations.16,23,24
Regulated necrosis may occur through necroptosis, ferroptosis, pyroptosis, or mitochondria permeability transition-regulated necrosis (MPT-RN) (Fig. 1).25–27 Necroptosis is the best characterized from a molecular point of view. Necroptosis requires the interaction of RIPK1 and RIPK3 kinases and MLKL phosphorylation under conditions in which caspase-8 is not active. A well-characterized example is cell death induced in tubular epithelial cells by the lethal cytokine cocktail TWEAK/TNFα/IFNγ. This proinflammatory cytokine cocktail promotes apoptosis. Caspase inhibition prevents apoptosis, but results in a higher rate of death through necroptosis.28,29 Necroptosis, characterized by a therapeutic effect of maneuvers that target molecular mediators of necroptosis, has been observed in experimental AKI induced by renal ischemia-reperfusion injury (IRI), rhabdomyolysis, and nephrotoxicity induced by cisplatin or crystals.29–33
 Apoptosis and necroptosis. TNF superfamily cytokines may activate both apoptosis and necrosis, inhibition of caspases promotes the occurrence of necroptosis. (B) Ferroptosis. (C) Pyroptosis. (D) Mitochondria permeability transition regulated necrosis (MPT-RN). Fer-1: ferrostatin-1; MPT-RN: mitochondria permeability transition-regulated necrosis; Nec-1: necrostatin-1; SfA: sanglifehrin A; zVAD: pan-caspase inhibitor z-VAD-fmk.")
Key molecular pathways in apoptosis and in diverse regulated necrosis forms. (A) Apoptosis and necroptosis. TNF superfamily cytokines may activate both apoptosis and necrosis, inhibition of caspases promotes the occurrence of necroptosis. (B) Ferroptosis. (C) Pyroptosis. (D) Mitochondria permeability transition regulated necrosis (MPT-RN). Fer-1: ferrostatin-1; MPT-RN: mitochondria permeability transition-regulated necrosis; Nec-1: necrostatin-1; SfA: sanglifehrin A; zVAD: pan-caspase inhibitor z-VAD-fmk.
Ferroptosis is characterized by an iron-dependent increase in lipid peroxidation resulting from glutathione depletion, and is inhibited by ferrostatin-1.34 Ferroptosis, defined by its response to ferrostatin-1 or other inhibitors, has been observed in different models of AKI such as renal IRI, oxalate nephropathy and folic acid overdose-induced AKI.35,36 Molecules that activate ferroptosis include erastin and RSL3. Erastin inhibits the antiporter system X−c, thus reducing the import of cystine into cells, which results in reduced glutathione levels. RSL3 inhibits glutathione peroxidase 4 (GPX4). GPX4 is a selenoprotein enzyme, which uses reduced glutathione to catalyze the reduction of hydrogen peroxide, organic hydroperoxides and lipid hydroperoxides inside biological membranes, protecting cells against oxidative damage. GPX4−/− mice develop spontaneous AKI, but the role of GPX4 in established preclinical models of AKI has not been studied.37 Interestingly ferroptosis triggered synchronized cell death in all cells in a single tubule.35 This may explain observations of multiple death cells in a single tubule with relative preservation of adjacent tubules, even in CKD.38 Ferroptosis may also modulate leukocyte function.39 Thus, T cell ferroptosis prevents immunity to infection, but the impact on kidney disease remains unexplored.40
Pyroptosis is a highly inflammatory, caspase-dependent type of cell death, and occurs mainly in macrophages and dendritic cells.26,41 During pyroptosis, the presence of danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) triggers the formation of a multiprotein complex called the inflammasome that activates caspase-1 and caspase-11. These caspases process pro-IL-18 and pro-IL-1β to mature proinflammatory cytokines that accumulate in the intracellular compartment. Cleavage of gasdermin-D by caspases leads to rupture of the plasma membrane, cell death and release of IL-1β and IL-18.42 Regarding the kidney, uromodulin and crystals promote macrophage inflammasome activation and pyroptosis.43 The occurrence of pyroptosis in tubular epithelial cells has been questioned, given their reduced capacity to release IL-1β.44 It was suggested that pyroptosis characterized by increased caspase-1 expression and IL-1β generation, may occur in renal tubular cells during renal IRI.45 However, the functional contribution of renal cell pyroptosis to IRI-induced AKI was not probed. Mice deficient for different component of inflammasome were used to demonstrate a role of the inflammasome in various experimental models of renal injury, but it is not clear the specific role of intrinsic renal cells in inflammasome activation.46
MPT-RN is another form of regulated necrosis also known as Cyclophilin-D (CypD)-mediated necrosis. This pathway is characterized by mitochondria dysregulation by the formation of the mitochondrial permeability transition pore (MPTP). CypD is key in opening the MPTP, but the composition of this pore is controversial.16,47 It was suggested that p53 and ATP synthase complex are involved. From a therapeutic point of view, MTP-RN has clinical relevance since CypD-KO mice were protected from renal IRI and from cisplatin nephrotoxicity.29,48
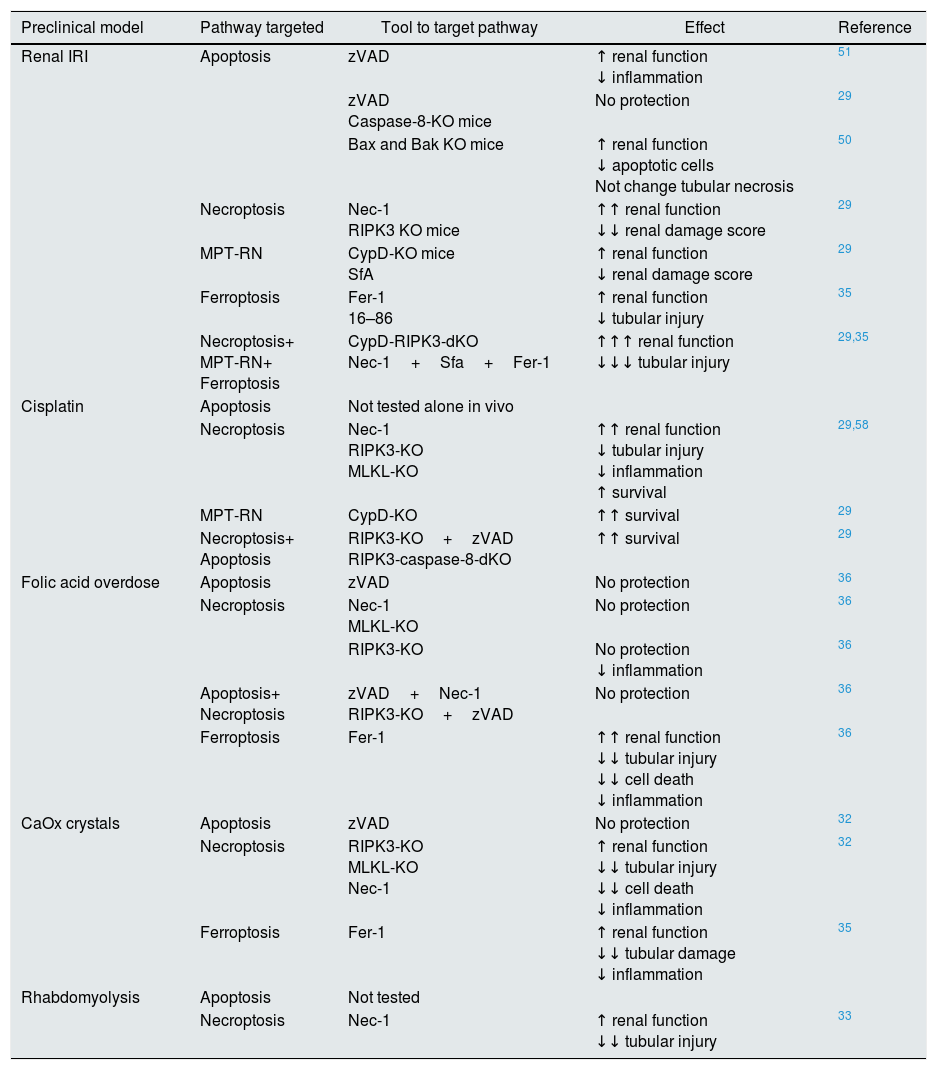
Targeting cell death in experimental AKIIn recent years, various preclinical functional studies have demonstrated that regulated necrosis plays a key role in AKI (Fig. 2) and new therapeutic targets have been proposed (Table 1).

Regulated necrosis in kidney disease. Different pathways of cell death are activated and contribute to kidney injury, based on intervention studies in vivo in preclinical models of AKI. Ferroptosis and necroptosis are the forms of cell death involved in most specific etiologies of AKI. IRI: ischemia-reperfusion injury.
Targeting regulated cell death in experimental AKI.
| Preclinical model | Pathway targeted | Tool to target pathway | Effect | Reference |
|---|---|---|---|---|
| Renal IRI | Apoptosis | zVAD | ↑ renal function ↓ inflammation | 51 |
| zVAD Caspase-8-KO mice | No protection | 29 | ||
| Bax and Bak KO mice | ↑ renal function ↓ apoptotic cells Not change tubular necrosis | 50 | ||
| Necroptosis | Nec-1 RIPK3 KO mice | ↑↑ renal function ↓↓ renal damage score | 29 | |
| MPT-RN | CypD-KO mice SfA | ↑ renal function ↓ renal damage score | 29 | |
| Ferroptosis | Fer-1 16–86 | ↑ renal function ↓ tubular injury | 35 | |
| Necroptosis+ MPT-RN+ Ferroptosis | CypD-RIPK3-dKO Nec-1+Sfa+Fer-1 | ↑↑↑ renal function ↓↓↓ tubular injury | 29,35 | |
| Cisplatin | Apoptosis | Not tested alone in vivo | ||
| Necroptosis | Nec-1 RIPK3-KO MLKL-KO | ↑↑ renal function ↓ tubular injury ↓ inflammation ↑ survival | 29,58 | |
| MPT-RN | CypD-KO | ↑↑ survival | 29 | |
| Necroptosis+ Apoptosis | RIPK3-KO+zVAD RIPK3-caspase-8-dKO | ↑↑ survival | 29 | |
| Folic acid overdose | Apoptosis | zVAD | No protection | 36 |
| Necroptosis | Nec-1 MLKL-KO | No protection | 36 | |
| RIPK3-KO | No protection ↓ inflammation | 36 | ||
| Apoptosis+ Necroptosis | zVAD+Nec-1 RIPK3-KO+zVAD | No protection | 36 | |
| Ferroptosis | Fer-1 | ↑↑ renal function ↓↓ tubular injury ↓↓ cell death ↓ inflammation | 36 | |
| CaOx crystals | Apoptosis | zVAD | No protection | 32 |
| Necroptosis | RIPK3-KO MLKL-KO Nec-1 | ↑ renal function ↓↓ tubular injury ↓↓ cell death ↓ inflammation | 32 | |
| Ferroptosis | Fer-1 | ↑ renal function ↓↓ tubular damage ↓ inflammation | 35 | |
| Rhabdomyolysis | Apoptosis | Not tested | ||
| Necroptosis | Nec-1 | ↑ renal function ↓↓ tubular injury | 33 |
CaOx: calcium oxalate. IRI: ischemia-reperfusion injury. Fer-1: ferrostatin-1; MPT-RN: mitochondria permeability transition-regulated necrosis; Nec-1: necrostatin-1; SfA: sanglifehrin A; zVAD: pan-caspase inhibitor z-VAD-fmk.
Transient renal ischemia followed by reperfusion is a cause of AKI, and death of tubular proximal cells is a key event. There is evidence of tubular cell apoptosis, such as activation of caspases, expression of pro-apoptotic proteins and typical apoptotic morphology.49 Bax and Bak conditional knockout mice were protected from renal IRI, but only partially, suggesting that other cell death pathways are activated.50 The effect of the pan-caspase inhibitor zVAD on experimental renal IRI is controversial. In one report, zVAD reduced serum urea levels and prevented inflammation, but the effect on kidney histology was not reported.51 In another report, zVAD did not protect from renal dysfunction or histological damage.30
By contrast, pre-treatment with the necroptosis inhibitor Necrostatin-1 (Nec-1) decreased the tubular histological injury score and serum urea and creatinine,30 suggesting that necroptosis may be a key pathway in renal IRI. In this line, mice deficient in the necroptosis regulatory protein RIPK3 were also protected from IRI.29 However, inhibition of necroptosis only offered a partial protection, suggesting that other pathways contribute to renal injury. Accordingly, MPT-RN has also been implicated in renal IRI. Chemical (e.g. sanglifehrin A, SfA) or genetic (e.g. CypD-ko) inhibition of CypD ameliorated renal injury after IRI.29 Moreover, renal injury was milder in CypD-RIPK3 double ko mice than in CypD-ko or RIPK3-ko mice. The same results were obtained with the combination of Nec-1 and the MPT-RN inhibitor SfA which showed a major therapeutic effect compared with the use of only one compound.29
Ferroptosis also contributes to experimental renal IRI. Both Fer-1 and a more stable analog, 16–86, improved renal function and reduced kidney damage after IRI.35 Furthermore, combination treatment against necroptosis, MPT-RN and ferroptosis offered a stronger protection.35
Thus, regulated necrosis is a key process in renal IRI. At least, three independent regulated necrosis pathways are involved, and inhibition of the three individual pathways was required for optimal protection. However, it is yet unclear whether individual pathways contribute to the loss of specific cell types or what factors result in the activation of either of these pathways and not the other, or whether all are activated simultaneously in the same cell. In this line, it is important to note that the effect of necroptosis inhibition is lower in isolated tubules, suggesting that tubular epithelium is not the only cell type that develops necroptosis. However, proximal tubular cells may undergo ferroptosis, suggesting that ferroptosis may be the principal contributor to tubular cell death after IRI.37 IRI occurs during renal transplantation, as discussed in more detail below under necroinflammation, and preclinical IRI studies may be relevant for kidney graft preservation.
Toxic and crystal-induced AKINephrotoxic agents and crystal deposition are frequent causes of AKI. Drug-induced nephrotoxicity accounts for up to 60% of hospital acquired AKI. Crystal deposition may result from drugs, minerals or metabolites.52,53 Most of these drugs induce renal cell death and understanding of the toxic mechanisms will provide useful information to develop drugs with therapeutic benefits and with reduced side effects.
Cisplatin nephrotoxicityCisplatin is an effective chemotherapy drug to treat numerous solid tumors but is associated with nephrotoxicity.54 Features of apoptosis have been observed in cisplatin nephrotoxicity.55 There is also evidence that cisplatin may induce apoptosis or necrosis in a dose-dependent fashion. In human tubular cells, low doses of cisplatin induce apoptosis, while at higher doses cell death shows features of necrosis.56 In cisplatin treated-tubular cells combined therapy against necroptosis (Nec-1) and apoptosis (zVAD) offered more protection than individual treatment.57 In vivo, Nec-1also prevented renal injury, and the combined inhibition of apoptosis and necroptosis prolongs survival of mice, suggesting that apoptosis also contributes to renal injury in nephrotoxicity by cisplatin.29,58 Moreover, RIPK3 or MLKL deletion also ameliorated cisplatin nephrotoxicity.58 However, additional pathways may be involved, since genetic deletion of both RIPK3 and MLKL only partially prevented renal damage and did not diminish cell death measured with TUNEL staining. In this line, MPT-RN may be also involved in cisplatin nephrotoxicity since it was observed that CypD-KO mice showed prolonged survival compared with WT mice.29
Folic acid overdose-induced AKI:Accidental folic acid overdose causes AKI in humans and experimental folic acid-induced AKI (FA-AKI) in mice is used to explore the pathophysiology of AKI.59,60 It is thought that folic acid precipitates inside tubules. Similar to cisplatin nephrotoxicity, changes in the expression of apoptotic regulatory proteins and caspase 3 activation are observed.61–63 However, pre-treatment with caspase inhibitors did not protect from FA-AKI, excluding a direct or key role of apoptosis, at least in the early phase of injury.36 Furthermore, necroptosis inhibition did not offer protection.36 By contrast, ferroptosis inhibition with Fer-1 improved renal function and histological tubular injury and decreased cell death.36 Moreover, ferroptosis inhibition also prevented inflammation associated to FA-AKI. This result suggests that ferroptosis is responsible for initial cell death in FA-AKI, triggering an inflammatory response and, maybe, activating other types of cell death in later phases of AKI. Further studies will be needed to clarify this.
While in RIPK3-ko mice renal function was not preserved, the inflammatory response was reduced, suggesting that RIPK3 may mediate inflammation during AKI independently from cell death modulation.36 This is different from the effect of necrostatin-136 and consistent with previous reports where RIPK3 absence diminished inflammation in experimental toxic colitis and in murine arthritis.64,65
Calcium oxalate nephropathy:Different crystals may cause kidney injury through shared molecular and cellular mechanisms.66 Calcium oxalate (CaOx) nephropathy is characterized by renal interstitial inflammation and renal failure.67,68 Inflammation resulting from inflammasome activation in immune cells locally in the kidney plays a key role. However, tubular cells can contribute to promote inflammation, since they internalize CaOx leading to cell death by necrosis and release of DAMPs such as ATP that activate the inflammasome in immune cells.68,69 Cultured tubular cells exposed to CaOx develop features of necrosis (propidium iodide uptake) but not of apoptosis (e.g. DNA fragmentation and annexin V binding).69 Moreover, necroptosis inhibition prevented CaOx-induced cell death in vitro and improved oxalate nephropathy in vivo, while apoptosis inhibition was not protective.68 A different report also implicated ferroptosis in CaOx-induced nephropathy in vivo.35 However, the relative contribution of each pathway is unknown, and a head-to-head comparison as well as combination treatment against both ferroptosis and necroptosis would be necessary to discern this.
Necroinflammation in kidney diseaseInflammation is another important feature of AKI and CKD. Both renal intrinsic cells and inflammatory cells contribute to inflammation during kidney injury.70,71 Kidney epithelial cells, such as tubular cells in AKI or podocytes in glomerular injury, release mediators of inflammation in response to stress or cell death that in turn recruit and activate inflammatory cells.72–75 Recent interest has focused in the causal relationship between regulated necrosis and inflammation and the term necroinflammation has been coined to describe this relationship.76,77 Necrotic cells release DAMPs and alarmins that activate immune renal cells and parenchymal cells leading to amplification of cell death and to the recruitment of immune responses.24 Both the innate and the adaptive immune system are recruited and activated in response to kidney injury and contribute to amplification of injury after the initial insult as has been clearly demonstrated in AKI. Monocytes are recruited by damaged tubular cells and differentiate into inflammatory macrophages that secrete cytokines and chemokines causing additional renal inflammation and damage.78 Moreover, there is also infiltration by neutrophils and natural killer cells and activation of dendritic cells.79,80 The adaptive immune system also contributes to AKI since T cell deficient (nu/nu) mice or CD4 deficient mice are protected from renal IRI and cisplatin nephrotoxicity,81,82 but the specific role of T lymphocytes in kidney injury beyond autoimmune disease and transplant rejection is not clear. Inflammation and immune cells can also play a role in recovery from kidney injury. In late stages of injury macrophages may differentiate to the M2 phenotype that resolves inflammation and promotes tissue recovery.78,83 Regulatory T cells (Tregs) also have a renoprotective role in kidney injury and, specifically in AKI. In fact, Tregs suppress inflammation by secreting anti-inflammatory cytokines and promote tubular proliferation.79,84,85
From the 90s apoptosis was considered the major pathway of cell death in AKI. This made it difficult to find a link between cell death and inflammation. However, the newly described contribution of regulated necrosis to AKI has opened a new field in the research on renal inflammation, since necrosis does promote inflammation. In necroinflammation, necrotic cells release DAMPs and alarmins,86 which have immunostimulatory properties and activate innate immune responses and nonimmune cells, such as parenchymal renal cells,87 triggering inflammation, which subsequently can generate more cell death by different mechanisms, in an auto-amplification loop of necrosis and inflammation that drives to acute organ dysfunction, organ failure, or even a systemic inflammatory response syndrome which can lead to multiple organ failure. DAMPs and alarmins include molecules known to contribute to renal injury including HMGB1, IL-33, histones and uromodulin.76,88 Furthermore, the inflammatory response elicited by regulated necrosis is highly immunogenic and could contribute to trigger acute and chronic rejection.88 In this regard, therapeutic approaches that limit IRI in renal grafts could decrease the incidence and severity of rejection in addition to limiting delayed graft function.89
FA-AKI could represent a good model to study necroinflammation because ferroptosis promotes the expression of Fn14 and kidney inflammation. TWEAK activation of Fn14 in an inflammatory milieu triggers cell death by apoptosis or necroptosis.28,29,36,90 In this regard, TWEAK targeting was nephroprotective and decreased inflammation and its consequences both in FA-AKI and in other forms of kidney disease.14,91,92 Moreover, IL-33 is also upregulated in response to ferroptosis during FA-AKI and could activate innate responses in lymphocytes.36
Diagnostic and therapeutic implicationsPreclinical studies have identified several pathways resulting in regulated necrosis that can be successfully modulated in AKI in vivo by drugs or interventions targeting the molecular pathways leading to regulated necrosis. Evidence has been obtained both for the potential contribution of different forms of regulated necrosis to the same model of AKI and for differences in the regulated necrosis forms relevant for diverse forms of AKI. Clinical development of drugs targeting regulated necrosis should be ideally associated to companion diagnostics developments that allow the early identification of the occurrence of diverse forms of cell death, ideally before renal function has decreased. Thus, approaches targeting regulated necrosis in clinical trials may consist on prophylactic administration of one or several compounds targeting one or several regulated necrosis pathways prophylactically in high risk situations such as patients undergoing heart surgery or in preservation solutions used for kidney grafts. The potential benefits may include prevention or decreasing the severity of AKI and, in the kidney transplantation context, reduce the immunogenicity of the graft. As an alternative, the drugs may be tested in patients that are monitored by companion diagnostics kits, aiming at identifying in urine features of ongoing regulated necrosis, and therapy started once the earliest features of kidney regulated necrosis are detected. Eventually the diagnosis effort of AKI may shift from identifying a cause and assessing the stage of injury to identifying which form/s of regulated necrosis are active in that particular patient and treating accordingly.
ConclusionsIn the nineties, the description of the molecular regulation of apoptosis raised hopes of therapeutic interventions in diseases conditions characterized by massive cell death, such as AKI. Unfortunately, the hype did not materialize into changes in clinical practice. However, the description of a new wave of regulated cell death modalities and early preclinical evidence of success in nephroprotection by targeting necroptosis, ferroptosis, pyroptosis, and/or MPT-RN, has raised again hopes of novel breakthroughs to prevent both renal cell death and necroinflammation. It is likely that in the future several forms of cell death are targeted simultaneously. The clinical applications may range from AKI, the condition most studied to date given the magnitude of cell death and the convenient logistics, to CKD or kidney graft preservation. Further studies are needed to define which specific forms of cell death should be targeted in each condition or whether a standardized anti-cell death cocktail targeting several cell death modalities may be applied to diverse clinical conditions. Agents better suited for clinical use should be developed or ideally, drugs that have already proved safe should be repurposed in order to accelerate clinical translation.93
Key points- ∘
Inhibitors of apoptosis did not lead to new therapeutic approaches in clinical practice.
- ∘
Unlike apoptosis, regulated necrosis promotes an inflammatory response (necroinflammation) which can explain the relation between cell death and inflammation during renal injury.
- ∘
Of interest to kidney transplantation, necroinflammation is immunogenic.
- ∘
The description of new pathways of regulated necrosis and early preclinical evidence of success in nephroprotection by targeting these pathways, have raised hopes of novel therapeutic approaches to prevent both renal cell death and inflammation.
- ∘
Further studies with specific inhibitors of regulated necrosis are needed to discern the specific pathway activated in each kidney disease.
The authors declare that they have no conflicts of interest.
Work by the authors has been funded mainly by a grant from the Spanish Society of Nephrology to ABS. Additional grants: FIS PI15/00298, PI16/02057, PI16/01900, PI13/00047, PI14/0041, CP14/00133, CP12/03262, ISCIII-RETIC REDinREN RD12/0021 and RD16/0009 FEDER funds, EUTOX, FRIAT-IRSIN, SENEFRO, CYTED IBERERC. Fundacion Conchita Rabago to DM-S, Programa Intensificación Actividad Investigadora (ISCIII) to AO, Miguel Servet to MDS-N and ABS, and Consejería de Educación, Juventud y Deporte (CAM and FSE) to MF-B.



