

Distal renal tubular acidosis (DRTA) is a rare disease resulting from a failure in the normal urine acidification process at the distal tubule and collecting duct level. It is characterised by persistent hyperchloremic metabolic acidosis, with a normal anion gap in plasma, in the presence of high urinary pH and low urinary excretion of ammonium.
To date, 5 genes whose mutations give rise to primary DRTA have been described. Alterations in the ATP6V1B1 and ATP6V0A4 genes are inherited recessively and are associated with forms of early onset and, in many cases, with neurosensorial deafness. Pathogenic variants in the SLC4A1 gene are habitually inherited dominantly and give rise to milder symptoms, with a later diagnosis and milder electrolytic alterations. Nonetheless, evolution to nephrocalcinosis and lithiasis, and the development of chronic kidney disease in the medium to long term has been described in a similar manner in all 3 groups. Lastly, recessive forms of DTRA associated to mutations in the FOXI1 and WDR72 genes have also been described.
The clinical management of DTRA is based on bicarbonate or citrate salts, which do not succeed in correcting all cases of the metabolic alterations described and, thus, the consequences associated with them. Recently, a new treatment based on slow-release bicarbonate and citrate salts has received the designation of orphan drug in Europe for the treatment of DTRA.
La acidosis tubular renal distal (ATRD) es una enfermedad rara que se debe al fallo del proceso normal de acidificación de la orina a nivel tubular distal y colector. Se caracteriza por una acidosis metabólica hiperclorémica persistente, con anión gap normal en plasma, en presencia de un pH urinario elevado y baja excreción urinaria de amonio.
Se han descrito hasta el momento 5 genes cuyas mutaciones dan lugar a ATRD primaria. Las alteraciones de los genes ATP6V1B1 y ATP6V0A4 se heredan de forma recesiva y están asociadas a formas de inicio más precoces y con sordera neurosensorial en muchos casos. Las variantes patogénicas en el gen SLC4A1 se heredan habitualmente de forma dominante y dan lugar a cuadros más leves, con un diagnóstico más tardío y alteraciones electrolíticas menores. Sin embargo, la evolución a nefrocalcinosis y litiasis, y el desarrollo de enfermedad renal crónica a medio-largo plazo se ha descrito de forma similar en estos 3 grupos. Por último, se han descrito también formas recesivas de ATRD asociadas a mutaciones en los genes FOXI1 y WDR72.
El manejo clínico de la ATRD se basa en sales de bicarbonato o citrato, que no logran corregir en todos los casos las alteraciones metabólicas descritas y, por lo tanto, las consecuencias asociadas a ellas. Recientemente, un nuevo tratamiento basado en sales de bicarbonato y citrato de liberación prolongada ha recibido la denominación de medicamento huérfano en Europa para el tratamiento de la ATRD.
- -
Hereditary dRTA is caused by mutations in genes that encode transporters that regulate acid-base balance at the distal tubular level.
- -
Mutations in the ATP6V0A4 and ATV6V1B1 genes are associated with earlier forms of dRTA, and it is frequently accompanied by the development of sensorineural deafness since childhood.
- -
The evolution to chronic kidney disease is common in all hereditary forms of dRTA and this is due to various factors, nephrocalcinosis/kidney stones, chronic hypokalemia and the frequent occurrence of acute pyelonephritis. In addition, poor metabolic control may worsen the evolution of kidney function.
- -
Traditional treatment with bicarbonate or citrate salts does not achieve optimal metabolic control in all cases.
Distal renal tubular acidosis (dRTA) is a rare disease with an estimated incidence <1: 100,000.1 Also known as type 1 RTA or classic RTA, it is characterized by persistent hyperchloremic metabolic acidosis, with a normal anion gap in plasma, in the presence of high urinary pH and low urinary ammonium excretion. dRTA is due to the failure of the type A renal intercalated cells of the collecting duct to produce a normal acidification of the urine, which is due to a dysfunction in any of the transporters involved in this process.1,2
EtiologyDTRA can be acquired or inherited. Acquired forms usually occur in adults and may be caused by drugs (especially antimicrobial, anti-inflammatory, diuretics and antivirals),3 autoimmune diseases syndrome (Sjögren, systemic lupus erythematosus, primary biliary cholangitis, cholangitis sclerosing primary, autoimmune hepatitis and autoimmune thyroiditis)4,5 or may be secondary to uropathies or kidney transplantation. It is not known with detail the mechanisms leading to these acquired forms. It may be caused by to alterations that are voltage dependent, lack of negative transepithelial difference in the distal lumen or defects of gradient.6
Hereditary forms are most frequent in pediatric patients, and is due to alterations in the genes that encode or control the encoding of the channels involved in urinary acidification in the distal and collecting tubules levels (Fig. 1).2,6,7 Currently, there are 5 recognized genes whose mutations can give rise to dRTA: ATP6V0A4, ATP6V1B1, SLC4A1, FOXI1 and WDR7.
1 or band 3 protein.")
Acid–base transporters involved in the secretion of H+ and reabsorption of HCO3– in the intercalated alpha cells of the collecting duct. Transport of H+ to the tnbular lumen is mediated by the vacuolar H+-ATPase, which actively transfers this proton across the luminal membrane. H+ can also be secreted thanks to a second ATPase, the H+/K+-ATPase, which exchanges H+ for K. Bicarbonate, formed intracellularly by the action of CAII, leaves the cell through the basolateral membrane, via the transport Anion exchange protein (AE)1 or band 3 protein.
Defects in the activity of the H+ ATPase (V-ATPase ) pump cause most primary cases of dRTA. H+ -ATPase is a highly conserved proton pump that is expressed in intercalated alpha cells and is made up of 2 domains, V1 and V0. The B1 subunit, encoded by the ATP6V1B1 gene, is part of the V1 domain that captures protons from the cell cytoplasm. This subunit is also expressed in the cells of the inner ear and the endolymphatic sac, among others.8,9 The A4 subunit, encoded by the ATP6V0A4 gene, is part of the V0 transmembrane domain involved in the translocation of protons through the cell membrane and is expressed in the kidney, inner ear and epididymis.9,10
This type of dRTA follows an autosomal recessive pattern of inheritance. Among the most frequent mutations in these genes are nonsense, frameshift or splice-site mutations, which are expected to alter the encoded protein, whereas missense mutations have only been described in a few patients.8,11–13 Experiments in cell culture models have shown that most of the mutations identified in this B1 subunit cause a dysfunction or alteration in the assembly of the protein complex that forms the V-ATPase pump.14 Mutations in the A4 subunit can affect the binding of these 2 subunits, leading to an incorrect assembly of the V1 and V0 domains, forming a structurally and functionally defective V-ATPase.15
In Europe, the most frequent mutations are in the ATP6V1B1 and ATP6V0A4.16 In our experience, most patients in northern Spain have mutations in the ATP6V0A4 gene. This is consistent with previous studies that report a higher frequency of mutations in this gene compared to the ATP6V1B1 gene in the Spanish and European population.6,17 Among them, the presence of the c.1691 + 2dup variant in different patients in our cohort is particularly noteworthy, as a reflection of a possible founder effect in this geographic region (data pending publication).
Mutations in the SLC4A1 geneThe SLC4A1 gene plays a crucial role in acid-base homeostasis since it encodes a chloride-bicarbonate exchanger, also known as AE1 or band 3 protein, responsible for the reabsorption of HCO3− together with the excretion of chloride.18 This protein is expressed in the plasma membrane of erythrocytes and the basolateral membrane of the renal intercalated alpha cells of the collecting tubules.19
The inheritance of this type of dRTA is complex, with forms of transmission both autosomal dominant, especially in Caucasians, and recessive, more common in Asians.20,21 The clinical manifestations are more severe in patients with recessive inheritance dRTA.22
Different types of mutations have been described, which in experimental studies cause intracellular retention of the mutated protein, reduction of its transport activity, misfolding and degradation, or even misdirection towards the lumen membrane.23,24 The most common recessive inherited mutation, G701D, causes dRTA that in some cases is associated with hemolytic anemia.16 Among Caucasian patients, the most common autosomal dominant inheritance mutation is R589H.17 The introduction of this mutation in mice leads to intercalated cell dysfunction with reduced expression of proton pumps.16,25 In mice, it has been observed that the complete absence of the AE1 exchanger causes severe metabolic acidosis, while the heterozygotes did not show any apparent defect.16
In our population there are few cases described of dRTA with defects in this channel and they usually have a milder phenotype with a late onset with respect to mutations in previously described genes.9,26,27
Other genes associated with distal renal tubular acidosisThe genes previously described only explain 70–80% of the cases of primary dRTA, which proves the existence of other candidate genes responsible for a significant number of cases.17,28
Pathogenic variants have been described recently in homozygous in gene FOXI1 as responsible for early onset of sensorineural hearing loss and autosomal recessive dRTA.29 FOXI1 is a key transcription factor for the regulation of various membrane transporters, including those necessary for normal acidification at the distal tubule and innerear (AE1, AE4 and various V-ATPase subunits ).30 Enerbäck et al.29 demonstrated that in patients with homozygous mutations with loss of function in this factor, there is a very limited DNA-binding activity of FOXI1, which probably results in an inability for the transactivation of some of these transporter proteins, which causes a syndrome severe deafness and acidosis.
Likewise, homozygous pathogenic variants in the WDR72 gene have recently been described as a cause of hereditary dRTA.31 It is believed that this gene could be involved in intracellular trafficking, affecting the direction of acid-base regulating proteins, such as the AE1 transporter isoform or V- ATPase, causing intracellular retention or misdirection.31 Mutations in WDR72 have been associated to amelogenesis imperfecta, which includes a large group of inherited diseases that affect the formation of dental enamel.32
Different studies in animal models have revealed new genes that could be involved in DRTA. Some have been identified in mice, but there is no evidence that they cause dRTA in humans. Recently, whole exome sequencing studies in patients with dRTA have revealed the ATP6V1C2 gene as a new gene responsible for recessive dRTA.33 This gene encodes the C subunit of V- ATPase and is mostly expressed in intercalated collecting duct cells.
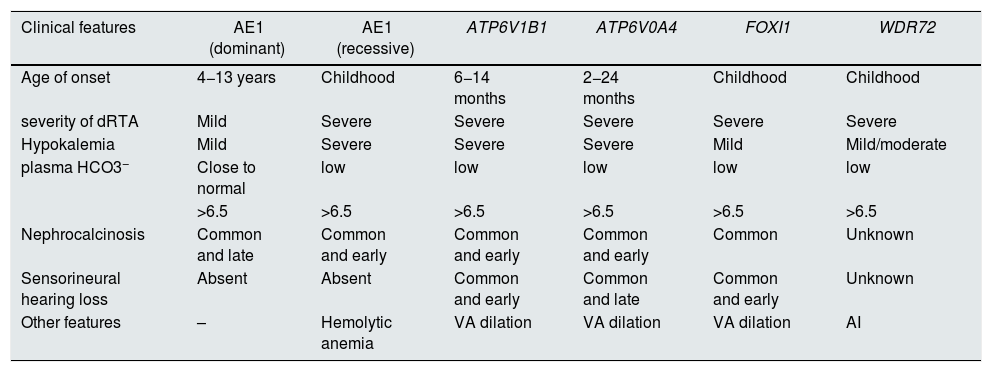
Clinical manifestations and genotype-phenotype correlationThe clinical manifestations of these genetic groups are common, although they present some peculiarity depending on the underlying causal gene (Table 1).
Differences in clinical characteristics between the main genetic groups causing dRTA.
| Clinical features | AE1 (dominant) | AE1 (recessive) | ATP6V1B1 | ATP6V0A4 | FOXI1 | WDR72 |
|---|---|---|---|---|---|---|
| Age of onset | 4−13 years | Childhood | 6−14 months | 2−24 months | Childhood | Childhood |
| severity of dRTA | Mild | Severe | Severe | Severe | Severe | Severe |
| Hypokalemia | Mild | Severe | Severe | Severe | Mild | Mild/moderate |
| plasma HCO3− | Close to normal | low | low | low | low | low |
| >6.5 | >6.5 | >6.5 | >6.5 | >6.5 | >6.5 | |
| Nephrocalcinosis | Common and late | Common and early | Common and early | Common and early | Common | Unknown |
| Sensorineural hearing loss | Absent | Absent | Common and early | Common and late | Common and early | Unknown |
| Other features | – | Hemolytic anemia | VA dilation | VA dilation | VA dilation | AI |
AI: amelogenesis imperfecta; ATRD: distal renal tubular acidosis; AV dilation: dilation of the vestibular aqueduct.
Clinical features at presentation. Clinical presentation in patients with hereditary dRTA usually begin in childhood with failure to thrive, repeated vomiting, polyuria, and episodes of acute decompensation generally coinciding with infectious pictures.17,28 Biochemical findings include a marked metabolic acidosis with a normal anion gap due to the absence of acids would count as anions, and a urine, with a pH generally greater than 6. The urinary anion gap is positive which reflects a of low urinary excretion of ammonium. The hypokalemia is common in these patients, it is present in a 30–50% of the patients; it is due to secondary hyperaldosteronism and to the defect of gradient in the distal tubule, and produces muscle weakness, constipation, inability to concentrate urine and, in extreme cases, paralysis and arrhythmias which can be fatal.6,34,35 In general, people with recessive DTRA (ATP6V0A4 and ATP6V1B1) have a more severe degree of metabolic acidosis and hypokalemia than those with dominant inheritance, who usually present a milder form of acidosis partially compensated.36,37 Polyuria secondary to a deficit in urinary concentration is very common in these patients and, although the underlying mechanisms have not been fully elucidated, probably it is caused by biochemical abnormalities associated, mainly to hypokalemia and hypercalciuria.38 In some patients, an associated transient proximal tubule dysfunction is observed at diagnosis, which may be confused with Fanconi syndrome.39,40 Occasionally, they can also present with hyperammonemia, which tends to resolve when acidosis is corrected.17,41
Regarding the age of onset, cases of recessive dRTA (ATP6V0A4 and ATP6V1B1) are associated with an earlier presentation of symptoms, generally at 6 and 24 months after birth.1,28 However, patients with a dominant dRTA (SLC4A1) have a later clinical onset, with a mean age at diagnosis between 4 and 13 years.1,9,28,36,37
Growth. dRTA is frequently diagnosed during the evaluation of growth defects in childhood, mainly with poor linear growth with normal weight.17 This is primarily attributed to the inhibitory effect of metabolic acidosis on growth hormone metabolism. Cases with mutations in the SLC4A1 gene tend to have less degree of growth retardation than recessive forms due to later presentation and milder metabolic acidosis.36 An adequate alkaline therapy tend to improve the growth, although some recent series show that the final height is frequently below the mean, regardless of the genetic group.1,17,42
Hearing loss. Due to the importance of proton secretion in endolymph for inner ear function, dRTA is also associated with sensorineural deafness. This occurs in individuals with mutations in ATP6V0A4 and ATP6V1B1, due to the simultaneous expression of V-ATPase in the kidney and ear, and in FOXI1. However, the frequency of deafness associated with mutations in ATP6V1B1 is much higher, being observed in around 90% of patients, compared to 35–50% of those with mutations in ATP6V0A4.1,28 Furthermore, there is a significant difference in the age of onset of deafness between these 2 groups. The detection of deafness in the first years of life is highly indicative of an underlying mutation in the ATP6V1B1 gene.28,36 Some patients also present other abnormalities of the auditory system, such as dilation of the vestibular aqueducts, which is generally bilateral, and they may suffer from dizziness.43,44 The cases described with pathogenic variants biallelic the gene FOXI1 present dRTA deafness and the dilated vestibular aqueduct.29
Skeletal manifestations. Metabolic acidosis causes the release of bicarbonate and phosphate from the bone, which act as alkaline buffers to restore physiological blood pH. This results in bone demineralization that can cause rickets in children and osteomalacia in adults. These abnormalities have been described, especially at diagnosis and in a variable manner, in some patients with dRTA.17,42,45
Hemolytic anemia. There are some cases reported, mostly in Southeast Asia, in pathogenic variants in the SLC4A1 gene, of recessive inheritance, that can give rise to dRTA and hemolytic anemia, generally in children.46 The importance of this fact is that these biallelic variants can produce morphological changes in erythrocytes and these, under conditions of metabolic acidosis, may undergo hemolysis. Alkaline therapy corrects anemia and reticulocytosis. These patients also respond to transfusions and ion therapy.42
Renal manifestations. Patients with dRTA very frequently and early develop nephrocalcinosis or lithiasis, due to the combination of hypercalciuria, hypocitraturia and a high urinary pH that favors the deposit of oxalate and calcium phosphate crystals. The hypocitraturia is virtually universal in these patients and is due to increased citrate reabsorption in the proximal tubule in response to systemic acidosis. The probability of developing nephrocalcinosis/lithiasis increases with the age of the patient and with the delay in the initiation of alkalizing treatment, that is, with a late diagnosis. Nephrocalcinosis is observed in 90–95% of patients, of any genetic group, and persists in most cases despite adequate therapeutic control. Adequate alkaline therapy usually prevents its progression, but does not reverse it.1,28,36 The frequency of calculi in patients with mutations in the SLC4A1 may be greater, possibly due to a delay in diagnosis caused by milder phenotype expression.1 Furthermore, patients with dRTA frequently present with recurrent acute pyelonephritis, associated with the presence of potentially obstructive stones and possibly hypercalciuria. Finally, many individuals develop spinal cysts in childhood or adulthood, which has been attributed to some extent to chronic hypokalemia. However, other tubulopathies with sustained hypokalaemia, such as Bartter syndrome, rarely present medullary cysts during its the evolution.17,47 The clinical relevance of these cysts is unknown as it was not found a correlation between the development of medullary cysts and the degree of nephrocalcinosis or the long - term deterioration of renal function.17
Incomplete distal renal tubular acidosisIncomplete dRTA cases have been described in patients who are heterozygous carriers of pathogenic variants in the ATP6V1B1 gene. These present mild renal acidification defects that do not lead to reduction in blood pH, but fail to adequately acidify the urine if stimulated. In these cases, functional tests to assess maximum urinary acidification capacity may be useful. These individuals frequently have associated hypercalciuria and hypocitraturia, and an elevated risk of kidney stones.42,48,49 Similarly, some dominant mutations in the AE1 transporter can give rise to incomplete dRTA.50
PregnancyWomen with dRTA do not present a priori fertility disorders associated with their disease. However, during pregnancy they may present metabolic complications associated with dRTA mainly hypokalemia and severe metabolic acidosis. In addition, hyperemesis gravidarum may cause electrolyte imbalances by increasing electrolyte losses and making oral supplementation difficult. Other complications to take into account are recurrent episodes of acute pyelonephritis, already common in patients with dRTA, and ureteral obstruction due to pre-existing kidney stones. The worsening of kidney function or proteinuria are not complications usually observed in pregnant women with dRTA, except in those cases with previous chronic kidney disease (CKD).16,35
Long-term evolution of kidney functionIn recent years, there have been published large series of patients with dRTA with confirmed molecular diagnosis evaluating the risk of CKD at the long term, describing CKD grade ≥2 in 30–80% of patients, without significant differences between the patients. 3 main genes involved in dRTA.1,17,28 Factors potentially involved in this progression are late diagnosis, with a greater number of episodes of decompensation and acute renal failure, nephrocalcinosis or nephrolithiasis particularly obstructive, repeated episodes of pyelonephritis, medullary cysts and persistent hypokalemia.21,28 Despite the fact that, as expected, the degree of CKD worsens with a longer evolution time, many patients already have mild CKD since adolescence.1,28
Despite the trend to CKD, the clinical evolution of patients with dRTA is not critical, particularly if the diagnosis is established early with the consequent correction of acidosis. Although CKD is common, end-stage kidney disease is rare. In many patients, adequate correction of acidosis and electrolyte disturbances is not achieved, reflecting the difficulties with the usual forms of treatment with supplements, both in tolerance and in maintaining therapeutic compliance. Adequate metabolic control can reduce the risk of developing CKD at the long term.1
Management of distal renal tubular acidosis. New treatmentsThe main objective in the management of dRTA is the correction of metabolic acidosis and the secondary and associated alterations, mainly hypokalemia, hypercalciuria and hypocitraturia. This is to ensure normal growth and development in childhood, avoid bone demineralization and associated rickets, and avoid CKD development factors in the medium-long term. The alkalizing treatment, however, does not succeed in modifying the onset or evolution of sensorineural deafness.
Correction of acidosis is carried out with bicarbonate or citrate salts, and the dose usually required for this is higher in the young child and decreases with the end of growth. Thus, in infants, alkali doses of up to 8 mEq/kg/day are required, later in children, doses of 3–4 mEq/kg/day are usually sufficient, and in adults, doses greater than 2–3 mEq/kg/day are generally not required. The use of citrate as an alkalinizing agent has the advantage of correcting the hypocitraturia associated with acidosis and of avoiding to some extent the development of associated nephrocalcinosis. The alkalizing agent is used in combination with sodium or potassium. The use of sodium makes sense especially in patients with polyuria and extravascular volume depletion, but it has the disadvantage of increasing calcium excretion and decreasing proximal bicarbonate reabsorption due to the volume overload that it may entail. The use of potassium also attempts to correct the associated hypokalemia.
The most important drawback of the use of alkalizing salts, especially citrate, is poor gastrointestinal tolerance. In addition, it is necessary to divide the treatment into several doses per day to maintain a constant acid-base homeostasis. On many occasions, adequate metabolic control is not achieved.1 Recently, the ADV7103 molecule, based on prolonged-release citrate and potassium bicarbonate (2 daily administrations), has been commercialized in order to improve therapeutic adherence and metabolic control in patients with dRTA. A phase 3 study has shown its efficacy in improving metabolic control and its safety with good gastrointestinal tolerance. This treatment has obtained in Europe the designation of orphan drug for the treatment of dRTA.
Some groups use amiloride as a potassium-sparing diuretic to optimize the treatment of hypokalaemia and to allow a reduction in potassium supplementation doses.2 Sometimes they have been used thiazides as hypercalciuria treatment, in order to prevent progression and nephrocalcinosis ERC.17 However, this treatment is not generally recommended as it potentially worsens potassium loss and there is no evidence that it improves kidney function in the long term.
To evaluate sensorineural hearing abnormalities, a standard audiogram should be performed to explore masked and unmasked air and bone conduction at different frequencies. Both magnetic resonance imaging and computed tomography can be used to diagnose dilatation of the vestibular aqueduct.16,43 Hearing devices (hearing aids or cochlear implants) and language teaching are essential to guarantee the normal intellectual development and social integration of these patients.16
Financial supportThe pharmaceutical company Advicenne (France) has provided funding for the study of patients with distal renal tubular acidosis, but has not participated in the preparation of this article (grant BC/A/19/039).
Conflict of interestsNo conflict of interest to declare
Please cite this article as: Gómez-Conde S, García-Castaño A, Aguirre M, Herrero M, Gondra L, Castaño L, et al. Acidosis tubular renal distal hereditaria: correlación genotípica, evolución a largo plazo y nuevas perspectivas terapéuticas. Nefrologia. 2021;41:383–390.



