



Primary distal renal tubular acidosis (dRTA) is a rare tubulopathy characterised by the presence of hyperchloremic metabolic acidosis. It is caused by the existence of a defect in the function of the H+ -ATPase located on the luminal side of the α-intercalated cells or the Cl − HCO3− (AE1) anion exchanger located on the basolateral side. Patients do not acidify the urine after acid overload (NH4Cl) or after stimulating H+ secretion by obtaining a high intratubular concentration of an anion such as chlorine (pH is measured) or HCO3- (urinary pCO2 is measured). We present a family with autosomal dominant dRTA produced by a heterozygous mutation in the SLC4A1 gene in which the two paediatric members showed a test of normal maximum urinary pCO2. Our hypothesis is that since the H + -ATPase is intact, at least initially, the stimulation induced by intratubular electronegativity to secrete H + could be effective, which would allow the maximum urinary pCO2 to be paradoxically normal, which could explain the onset, moderate presentation of symptoms and late diagnosis in patients with this mutation. This is the first documented case of a dominant dRTA in Mexico.
La acidosis tubular renal distal (ATRd) primaria es una tubulopatía poco frecuente caracterizada por la presencia de acidosis metabólica hiperclorémica. Está generada por la existencia de un defecto en la función de la H+-ATPasa situada en el lado luminal de las células α-intercaladas o del intercambiador de aniones Cl−-HCO3− (AE1) ubicado en el lado basolateral. Los pacientes no acidifican la orina tras una sobrecarga ácida (NH4Cl) o tras estimular la secreción de H+ mediante la obtención de una elevada concentración intratubular de un anión como cloro (se mide el pH) o HCO3− (se mide la pCO2 urinaria). Se presenta una familia con ATRd autosómica dominante producida por una mutación heterocigota en el gen SLC4A1 en la que los dos miembros en edad pediátrica mostraron una prueba de la pCO2 urinaria máxima normal. Nuestra hipótesis es que al estar intacta, al menos inicialmente, la H+-ATPasa, podría ser efectivo el estimulo inducido por la electronegatividad intratubular para secretar H+ lo que permitiría que la pCO2 urinaria máxima fuera paradójicamente normal, lo que pudiera explicar el inicio tardío, la presentación moderada de los síntomas y el diagnóstico en edades más avanzadas, en los pacientes con dicha mutación. Este es el primer caso documentado de una ATRd dominante en México.
Distal renal tubular acidosis (dRTA) (Butler-Albright disease, nephrocalcinosis infantum) is a clinical syndrome characterised by hyperchloraemic metabolic acidosis. It is caused by a defect in the urinary excretion of hydrogen ion (H+) in the distal portions of the nephron, which makes them unable to lower the urine pH below 6.1,2 It is a rare disease, which causes growth retardation, polyuria, hypercalciuria, hypocitraturia, nephrocalcinosis, potassium depletion and characteristic extrarenal manifestations in some of the types.2,3
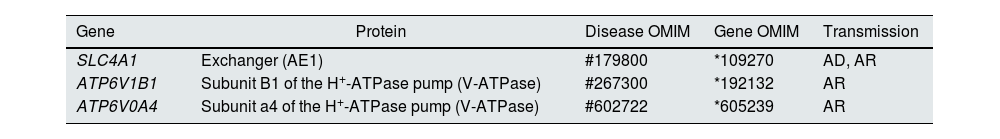
Distal tubular acidification of urine under normal conditions is summarised in Fig. 1. Vacuolar H+-ATPases (V-ATPase) are highly conserved proton pumps expressed in α-intercalated cells and are made up of two domains, V1 and V0. Defects in the activity of the H+-ATPase pump cause the majority of primary cases of autosomal recessive hereditary dRTA. They are due to mutations in the ATP6V1B1 and ATP6V0A4 genes (Table 1) that encode the B1 and A4 subunits of the H+-ATPase pump, respectively.4–6 Autosomal recessive dRTA associated with deafness can be caused by mutations in the FOXI1 gene that encodes a transcription factor necessary for the expression of at least four subunits of H+-ATPase (A1, B1, E2 and a4).7 A fourth gene, WDR72 (OMIM 613211), when mutated produces another variety of autosomal recessive dRTA. This gene seems to be involved in the intracellular trafficking of proteins that regulate the acid-base balance, causing their intracellular retention or misdirection.8,9 Extrarenal manifestations include: 1) patients with recessive dRTA caused by mutations in the ATP6V1B1 or ATP6V0A4 genes may frequently and constantly have sensorineural deafness in FOXI1 gene mutations, with early onset and associated with Pendred syndrome (ORPHA:705)8; and 2) patients with recessive dRTA caused by WDR72 gene mutations may develop amelogenesis imperfecta.
 reabsorption of the residual amount of bicarbonate (10%), which has not been recovered in more proximal areas of the nephron; b) titration of the divalent phosphate anion (HPO42–) with H+, which is transformed into monovalent phosphate anion (H2PO4−) or titratable acidity; c) accumulation of intraluminal ammonia (NH3), which take up H+ and forms ammonium (NH4+). The secretion of H+ and the titration of urinary buffers lead to acidification of the urine, with which pH values close to 4.5 can be reached under conditions of maximum stimulation of the process. This takes place in the α-intercalated cells in the late distal convoluted tubule, the connecting tubule and in the cortical and medullary collecting ducts. Distal secretion of H+ generates an equimolar amount of bicarbonate. For each hydrogen molecule excreted into the tubular lumen, a new bicarbonate molecule is generated intracellularly thanks to the action of intracytoplasmic carbonic anhydrase (CA type II), which is transferred to the blood through the Cl−-HCO3− anion exchanger (AE1). The α-intercalated cells secrete H+ via vacuolar ATPase (H+-ATPase), which actively transfers H+ across the luminal membrane, and H+-K+-ATPase, which exchanges H+ for potassium. The function of H+-ATPase is markedly influenced by the electronegativity generated in the tubular lumen by the simultaneous transport of Na+ in the principal cells of the collecting duct. The accumulation of NH3/NH4+ in the medulla generates a concentration gradient that helps it to cross through the basolateral membrane of the α-intercalated cells. NH3/NH4+ excretion requires at least two steps: basolateral entry and luminal excretion. Uptake from the NH3/NH4+ interstitium is carried out by several pathways including the Na+/K+/2Cl− transporter (NKCC1), Na+-K+-ATPase (in the case of these two transporters, NH4+ can be transported instead of potassium), RhCG (human Rhesus C glycoprotein) gas channels and hyperpolarisation-activated cyclic nucleotide-gated (HCN) cationic non-selective NH4+ channels). The luminal membrane has a high permeability for NH3. RhCG channels are present in the luminal membrane and in the basolateral membrane.")
Distal urine acidification under normal conditions. Urine acidification occurs in the distal and collecting tubules through three related processes: a) reabsorption of the residual amount of bicarbonate (10%), which has not been recovered in more proximal areas of the nephron; b) titration of the divalent phosphate anion (HPO42–) with H+, which is transformed into monovalent phosphate anion (H2PO4−) or titratable acidity; c) accumulation of intraluminal ammonia (NH3), which take up H+ and forms ammonium (NH4+). The secretion of H+ and the titration of urinary buffers lead to acidification of the urine, with which pH values close to 4.5 can be reached under conditions of maximum stimulation of the process. This takes place in the α-intercalated cells in the late distal convoluted tubule, the connecting tubule and in the cortical and medullary collecting ducts.
Distal secretion of H+ generates an equimolar amount of bicarbonate. For each hydrogen molecule excreted into the tubular lumen, a new bicarbonate molecule is generated intracellularly thanks to the action of intracytoplasmic carbonic anhydrase (CA type II), which is transferred to the blood through the Cl−-HCO3− anion exchanger (AE1). The α-intercalated cells secrete H+ via vacuolar ATPase (H+-ATPase), which actively transfers H+ across the luminal membrane, and H+-K+-ATPase, which exchanges H+ for potassium. The function of H+-ATPase is markedly influenced by the electronegativity generated in the tubular lumen by the simultaneous transport of Na+ in the principal cells of the collecting duct. The accumulation of NH3/NH4+ in the medulla generates a concentration gradient that helps it to cross through the basolateral membrane of the α-intercalated cells. NH3/NH4+ excretion requires at least two steps: basolateral entry and luminal excretion. Uptake from the NH3/NH4+ interstitium is carried out by several pathways including the Na+/K+/2Cl− transporter (NKCC1), Na+-K+-ATPase (in the case of these two transporters, NH4+ can be transported instead of potassium), RhCG (human Rhesus C glycoprotein) gas channels and hyperpolarisation-activated cyclic nucleotide-gated (HCN) cationic non-selective NH4+ channels). The luminal membrane has a high permeability for NH3. RhCG channels are present in the luminal membrane and in the basolateral membrane.
Basolaterally, only one type of autosomal dominant inheritance of dRTA has been described (Table 1) in most cases. It is caused by mutations in the SLC4A1 (OMIM 109270) gene, which encodes the two isoforms of the Cl−-HCO3 anion exchanger, the renal isoform also known as kAE1 (kidney anion exchanger 1) and the red blood cell isoform also known as band 3 protein (erythroid isoform or eAE1); kAE1 is responsible for HCO3− reabsorption along with Cl− excretion in α-intercalated cells (Fig. 1).10 This type of dRTA has been associated with less severe forms of clinical presentation, with late onset in childhood, adolescence and in adult patients, with less impact on growth compared to autosomal recessive inheritance.8
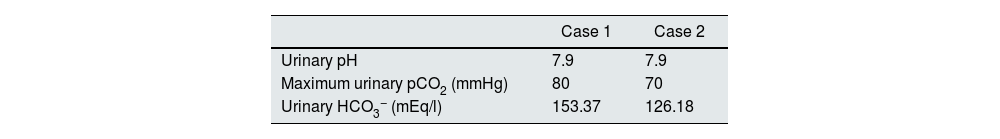
It has been reported that patients with dRTA do not acidify their urine after an acid overload (NH4Cl) or after H+ secretion being stimulated by a high intratubular concentration due to an anion such as chlorine (furosemide, Cl2Ca). Neither does their urinary pCO2 increased above 70 mmHg despite a high intratubular concentration of HCO3−.11 We present a family with autosomal dominant dRTA caused by a heterozygous mutation in the SLC4A1 gene, in which two of its paediatric members had a normal maximum urinary pCO2 test.
Case reportsCase 1This was a female patient aged four years three months. Birth weight 2800 g, length 48 cm and Apgar 8/9. History of craving for water and salt. Polyuria and nocturia as an infant. She was hospitalised 11 times for fever with no obvious focus. At the age of 20 months, she was diagnosed with herpes zoster and treated with aciclovir for seven days; subsequently, she developed diarrhoea, moderate dehydration and general flaccidity with the inability to walk. She was admitted to the Hospital General de Zona with suspected Guillain-Barré syndrome; weight 9.5 kg and height 82 cm (both in the 10th percentile). She was found to have hypokalaemia (2.5 mEq/l) and metabolic acidosis. After being started on intravenous treatment and the correction of her fluid/electrolyte imbalance, her strength improved and she was able to walk. However, the hyperchloraemic metabolic acidosis persisted. The clinical chemistry data are shown in Table 2 and the ultrasound findings in Fig. 2. A hearing test showed no evidence of sensorineural hearing loss. Her ophthalmological examination was normal. A urinary acidification test with sodium bicarbonate and acetazolamide was performed according to a previously described protocol (maximum urinary pCO2 test).11–13 The test was without complications, well tolerated and considered valid when bicarbonaturia greater than 80 mEq/l was found. A maximum urinary pCO2 of 80 mmHg was measured at 60 min (abnormal <70 mmHg) (Table 3). The patient was found to be a carrier of the same mutation as her mother. After three years of treatment with alkalis, her weight and height have improved (16.5 kg [47th percentile] and 103 cm [34th percentile], respectively). She is currently on treatment with potassium citrate (4.5 mEq/kg/day) and her hypercalciuria has remitted. Her blood pressure (BP) is normal (89/59 mmHg).
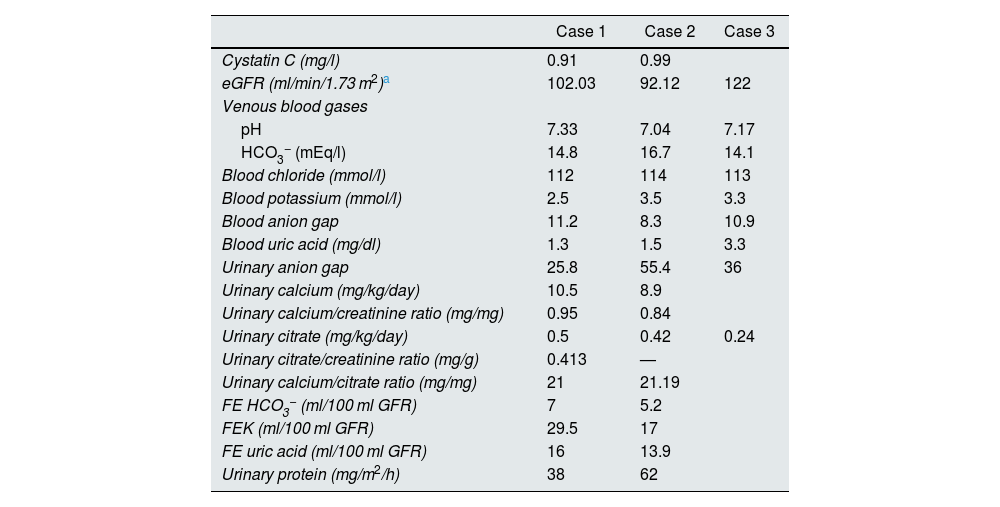
Blood and urine clinical chemistry tests.
| Case 1 | Case 2 | Case 3 | |
|---|---|---|---|
| Cystatin C (mg/l) | 0.91 | 0.99 | |
| eGFR (ml/min/1.73 m2)a | 102.03 | 92.12 | 122 |
| Venous blood gases | |||
| pH | 7.33 | 7.04 | 7.17 |
| HCO3− (mEq/l) | 14.8 | 16.7 | 14.1 |
| Blood chloride (mmol/l) | 112 | 114 | 113 |
| Blood potassium (mmol/l) | 2.5 | 3.5 | 3.3 |
| Blood anion gap | 11.2 | 8.3 | 10.9 |
| Blood uric acid (mg/dl) | 1.3 | 1.5 | 3.3 |
| Urinary anion gap | 25.8 | 55.4 | 36 |
| Urinary calcium (mg/kg/day) | 10.5 | 8.9 | |
| Urinary calcium/creatinine ratio (mg/mg) | 0.95 | 0.84 | |
| Urinary citrate (mg/kg/day) | 0.5 | 0.42 | 0.24 |
| Urinary citrate/creatinine ratio (mg/g) | 0.413 | — | |
| Urinary calcium/citrate ratio (mg/mg) | 21 | 21.19 | |
| FE HCO3− (ml/100 ml GFR) | 7 | 5.2 | |
| FEK (ml/100 ml GFR) | 29.5 | 17 | |
| FE uric acid (ml/100 ml GFR) | 16 | 13.9 | |
| Urinary protein (mg/m2/h) | 38 | 62 | |
eGFR: estimated renal glomerular filtration rate; FE: fractional excretion; FEK: fractional excretion of potassium; GFR: renal glomerular filtration rate; HCO3−: bicarbonate anion.

This was a male patient aged eight years 11 months with no relevant perinatal history (weight 2.9 kg, length 50 cm and Apgar 9/10). Cravings for water and salt from about two years of age. He had to be admitted to hospital three times before the age of 12 months with episodes of moderate dehydration. He was taken to the local Accident and Emergency department 24 times with fever. On some occasions it was classified as a respiratory tract infection, but in most cases no apparent cause was identified. He was investigated after his sister was diagnosed with dRTA. He weighed 18 kg (5th percentile) and his height was 111 cm (3rd percentile). He was found to have hyperchloraemic metabolic acidosis with normal anion gap, hypercalciuria, severe hypocitraturia (Table 2) and grade III nephrocalcinosis. His hearing test was normal. Maximum urinary pCO2 was also normal (Table 3). Both his weight (23 kg, 8th percentile) and his height (122 cm, 17th percentile) have also improved after three years of treatment with alkalis. He is on treatment with potassium citrate (5 mEq/kg/day). The genetic study showed the same mutation as his mother and sister.
In both children the morphology of their red blood cells was normal.
Case 3This was the 29-year-old mother of the patients, with a history of mother with diabetes and father with hypertension on treatment. She had an older brother with kidney stones and multiple episodes of flaccid paralysis. She was diagnosed after the investigations on her children. She had had a craving for water since childhood. She weighed 58 kg, was 157 cm tall and her blood pressure was 100/60 mmHg. Creatinine levels were 0.78 mg/dl (CrCl 122 ml/min), sodium 139 mEq/l, potassium 3.3 mEq/l and blood chloride 113 mEq/l. Venous blood gas analysis revealed metabolic acidosis (pH 7.17, HCO3− 14.1 mEq/l) with hypocitraturia (14 mg/24 h) and proteinuria 382 mg/24 h (Table 2). Her urinary pH was 7 and she had a positive urine anion gap (+36). Renal ultrasound revealed grade II-III medullary nephrocalcinosis (Fig. 3). A calculus was found in the lower segment of her right ureter and another 1.5 cm in size in her bladder. A genetic study was performed, analysing the genes ATP6V0A4, ATP6V1B1 and SLC4A1. A heterozygous pathogenic variant was found in exon 20 of the gene SLC4A1, c.2710_*12, p. (Tyr904_Val911delins68), resulting in a deletion of 39 bases (27 from the last exon and 12 from the 3′ UTR region), modifying the carboxy terminus of the protein (deletion of the last 8 amino acids and insertion of a new sequence of 68 amino acids). This variation, which is not described in the literature and not included in the database, is considered pathogenic, class 5 according to the American College of Medical Genetics and Genomics (ACMG) 2015 guidelines; the criteria used to classify it as class 5 are: PVS1, PM2, PM1 and PM4. This variation is not in the general population Genome Aggregation Database (gnomAD) (criterion PM2).

Ultrasound performed on the mother of the patients. Kidneys with heterogeneous echogenicity due to increased echogenicity of the renal pyramids consistent with grade II-III bilateral medullary nephrocalcinosis. Corticomedullary differentiation is poor, with signs of diffuse chronic nephropathy.
The peripheral blood samples were collected in EDTA tubes. Pure DNA was obtained using the QIAamp® DNA Blood MIDI Kit (Qiagen) in accordance with the manufacturer's instructions. The index case of this family was analysed with the gene panel using "next-generation sequencing" (previously described in Ashton E et al., Kidney Int, 2018 PMID: 29398133 and Hureaux M, Kidney Int, 2019 PMID: 31672324), which enabled analysis of the three major genes; FOXI1 was not analysed as it was not included in the panel at the time the study was conducted. Once the variation was identified, it was confirmed in the index case using the Sanger method and the other members of the family were also analysed with Sanger. The transcripts used to describe the variations were: ATP6V0A4: NM_130841.2; ATP6V1B1: NM_001692.3; and SLC4A1: NM_000342.3.
DiscussionWhen there is a genetic defect in the activity of the H+-ATPase pump, the metabolic acidosis caused by the defect in tubular secretion of H+ causes growth retardation, mobilisation of bone calcium14 and inhibition of tubular reabsorption of NaCl and calcium.15 Citrate is a very sensitive marker of metabolic acidosis.16 Reduction in the pH of the proximal tubular cell stimulates the activity of the dicarboxylate cotransporter, sodium-dependent dicarboxylate transporter 1 (NADC1), located in the apical membrane of the proximal tubule. NADC1 is a Na+-coupled electrogenic transporter mechanism for reabsorption (3 Na+ cations for every Cit2− anion). The consequence of increased tubular reabsorption is hypocitraturia. Once the citrate is reabsorbed, it is transported to the mitochondria where mitochondrial aconitase transforms it into isocitrate to enter the Krebs cycle17 in which citrate is converted to CO2 and H2O and three H+ are consumed, a reaction equivalent to generating bicarbonate. Hypercalciuria and hypocitraturia can increase the risk of nephrocalcinosis. As Na+ cannot be exchanged for H+ in the distal portions of the nephron, it needs to be exchanged with K+. Hypokalaemia can be a consequence of secondary hyperaldosteronism caused by volume contraction, which, in turn, is caused by salt loss and polyuria. Polyuria is the result of nephrocalcinosis, the aforementioned salt loss, body potassium depletion (hypokalaemic nephropathy)18,19 and, certainly, hypercalciuria itself.
What happens when the genetic defect affects the activity of the Cl−-HCO3− anion exchanger, as in our patients? Bicarbonate, formed intracellularly by the action of carbonic anhydrase, cannot leave the cell through the basolateral membrane, so the intracellular pH rises, which inhibits the activity of H+-ATPase. Patients with autosomal dominant dRTA therefore have urine with a pH above 6 in a situation of spontaneous acidosis20 or in acidification tests carried out with H+ stimulation, as in those performed with NH4CL.20,21 The lack of exchange with Cl− explains the hyperchloraemia typical of dRTA (Fig. 1).
However, as the H+-ATPase is intact, at least initially, the stimulus induced by intratubular electronegativity may be effective. Since the 1980s, it has been recognised that distal acidification capacity is dependent on anions22 and is stimulated by aldosterone.23 That explains at least two important points. Firstly, that clinical manifestation is later in patients with the autosomal dominant form, especially in childhood and adolescence, and is commonly a causal finding in adults; with a milder phenotype in adolescence or adulthood, with slightly low serum bicarbonate levels and with less involvement and compromise of weight and height, which tend to remain within appropriate percentiles for age.3,24 Some patients with heterozygous mutations in the SLC4A1 gene even have an incomplete dRTA phenotype, that is, without metabolic acidosis20,25,26 (in some patients with incomplete dRTA, the pCO2 test is normal25). In the course of evolution, it seems that the transcription of the different subunits that make up H+-ATPase must be progressively altered, causing the symptoms to become more evident. Secondly, the electronegativity caused by the high intratubular concentration of bicarbonate would stimulate H+-ATPase to secrete H+, which would result in normal maximum urinary pCO2 test results, as we found in our two patients.
Another important finding in the three cases we report here is hypouricaemia associated with hyperuricosuria, which has been described particularly in autosomal recessive forms in some cases of dRTA, as a consequence of endosomal dysfunction of proximal tubule cells induced by chronic intracellular acidosis or associated with nephropathy secondary to chronic hypokalaemia, which causes vacuolisation of proximal tubule cells.27,28
The very significant nephrocalcinosis patients develop is particularly striking. This has been reported in 94% of dRTA cases with mutations in the SLC4A1 gene, and is slightly more common, at 98%, in ATP6V0A4 mutations,24 most likely due to the severe hypocitraturia in our three cases, as well as the elevated hypercalciuria they had at diagnosis, which returned to normal once treatment potassium citrate was started.
The inheritance of this type of dRTA with mutations in the SLC4A1 gene is complex with, as mentioned above, autosomal dominant transmission, especially in Caucasians.21 However, a recessive form has also been described, which is more common in some tropical countries of Southeast Asia, particularly in Thailand, Malaysia, the Philippines and Papua New Guinea; in these countries, ovalocytosis due to heterozygous mutations of SLC4A1 (Southeast Asian ovalocytosis [SAO]) is common.29,30 Other mutations in this gene can also cause spherocytosis. Mutations that cause ovalocytosis or spherocytosis are responsible for morphological changes in red blood cells and haemolytic anaemia, and affect amino acids different from those affected in dRTA. Walsh et al.31 and other groups32 have tried to identify why dRTA is so common in Southeast Asia. Their published results suggest the hypothesis that the changes in red blood cells caused by these mutations could protect against malaria. In 1981, oval red blood cells from Melanesians living in Papua New Guinea were shown to be resistant to infection by malaria parasites (Plasmodium falciparum).33 Hereditary spherocytosis is a phenotypic and genetically heterogeneous condition. Some 20%–35% of cases are caused by mutations in the SLC4A1 gene.34 There have been reports of patients with biallelic mutations in this gene (for example, an SAO mutation and a distal acidosis mutation) who may suffer from haemolytic anaemia associated with dRTA.35–37
In 1999, Kaitwatcharachai et al. published the data of an adult patient with SAO in whom the urinary pCO2 test was normal.38 Years later, one of the co-authors of that paper reflected on the finding and suggested that some of the mutated kAE1 proteins would be expressed in the luminal membrane, which would produce “a net secretion of bicarbonate and an increase in pCO2 in the distal nephron”.39 To our knowledge, this hypothesis would not explain the increase in H+ secretion necessary to increase urinary pCO2.
In any event, the heterozygous mutation in exon 20 of the SLC4A1 gene found in our patients is the first time it has been described. Therefore, the return of urinary pCO2 to normal levels seen in our patients should not be specific to patients carrying the SAO mutation.
FundingWe thank patients and family for their valuable participation. This study was supported by the Fundación Para la Acidosis Tubular Renal Infantil Mexicana (FUNATIM; www.acidosistubular.unam.mx), Facultad de Medicina, Universidad Nacional Autónoma de México (UNAM).
Conflicts of interestThe authors declare that they have no conflicts of interest.



