




La Hiperglucemia como factor determinante de las complicaciones microvasculares de la diabetes
De manera característica, el hecho sin duda determinante en la fisiopatología de las complicaciones asociadas a la diabetes mellitas (DM) lo constituye la presencia de una situación de hiperglucemia crónica, siendo evidente que un mal control glucémico constituye un predictor independiente del desarrollo y progresión de la enfermedad renal asociada a la diabetes (ERAD), así como de otras complicaciones de la enfermedad1.
A pesar del reconocimiento de la hiperglucemia como condición necesaria y principal elemento determinante del desarrollo de la ND, seguimos sin conocer completamente los mecanismos íntimos por los cuales la hiperglucemia conduce a la lesión renal, aunque sí tenemos certeza de la participación fundamental de diversos procesos que confluyen para iniciar los cambios funcionales y estructurales a nivel renal (ej, hipertrofia glomerular, proliferación mesangial), y que van a conducir a una modificación de la hemodinámica corpuscular y la estimulación de procesos de proliferación e hipertrofia celulares. La modificación de diversas moléculas por el ambiente hiperglucémico, con la formación final de los productos avanzados de la glicosilación (AGEs), juega un papel fundamental. Asimismo, los niveles elevados de glucosa ejercen sus efectos tóxicos en el interior de las células a través de su incorporación por transportadores de glucosa, activándose una cadena enzimática de distintas reacciones que incluyen: formación de sorbitol, aumento de stress oxidativo, activación de la proteín kinsa C (PKC) y activación de la ruta de la hexosaminasa. Todas estas vías enzimáticas y metabólicas van a contribuir a la activación de citoquinas y de factores de crecimiento que participan de manera activa en la aparición y desarrollo de la ERAD2 (Figura 1).
La vía de los polioles y actividad de la aldosa-reductasa
La aldosa-reductasa (AR) es la primera enzima de la vía de los polioles, encargada de catalizar la reducción de una amplia variedad de compuestos carbonilo, incluyendo las hexosas. Se localiza a nivel citosólico y está presente en diferentes órganos y tejidos; así, la encontramos en el ojo (epitelio corneal, cristalino y pericitos retinales), en el riñón (podocitos, células mesangiales, epitelio tubular), y en los nervios periféricos (axones y células de Schwann)3. Esta enzima tiene muy baja afinidad por la glucosa, de forma que esta vía de metabolismo se encuentra usualmente inactiva, con una muy baja producción de sorbitol. Sin embargo, en presencia de hiperglucemia, y al aumentar la glucosa intracelular, se activa la AR con la producción creciente de sorbitol, lo que conlleva la consiguiente disminución de nicotinamida adenina dinucleótido fosfato (NADPH), iniciándose su propio proceso metabólico interfiriendo con la vía glicolítica normal. Así, en situación de hiperglucemia, el metabolismo de glucosa por esta vía es aproximadamente de un tercio del total. Posteriormente, el sorbitol, por acción de la sorbitol-deshidrogenasa, es metabolizado a fructosa. En todo este proceso tienen lugar cuatro fenómenos:
(i) producción de sorbitol, (ii) producción de fructosa, (iii) disminución del NADPH, y (iv) aumento del nicotinamida adenín dinucleótido reducido (NADH), cuyas implicaciones describiremos a continuación.
El sorbitol, al no difundir fácilmente a través de las membranas celulares, produce un aumento de la presión osmótica intracelular, con el potencial daño tisular por edema celular, aunque este mecanismo está lejos de dar lugar a una alteración osmótica definitiva. Sin embargo, y de manera particular en las fibras nerviosas, el aumento del sorbitol bloquea el contra-transportador Na+/Mioinositol, haciendo disminuir el mioinositol y los fosfoinositósidos intracelulares, lo que causa una depleción de diacilglicerol (DAG) y el subsiguiente freno en la actividad de la ATPasa Na+/K+, causando mayor edema axonal. La disminución del DAG ocurre exclusivamente en la neuropatía, pero no en la retinopatía ni la nefropatía asociadas a la diabetes, donde se observa un aumento de este compuesto.
La hipótesis más reciente para explicar parte del daño que se produce sugiere que la oxidación del sorbitol aumenta la relación NADH:NAD+, inhibiendo la actividad de la dehidrogenasa gliceraldehído trifosfato (GADPH) y aumentando la concentración de triosafosfato. Al elevarse las concentraciones de este compuesto se incrementa la formación de metilglioxal, un precursor de los AGEs, y de DAG, que activa la PKC4. El aumento de NADH favorece la síntesis de DAG, lo que ocurre tanto en el daño renal como en el retiniano, pero no en la afectación neuronal. Por otra parte, el consumo de NADPH favorece el estrés oxidativo al disminuir el cociente glutatión reducido/oxidado, lo cual acelera los procesos de glicosilación, así como aumenta la actividad de la vía de las pentosas, activando a su vez a la PKC5.
En resumen, la activación de la AR no sólo produce daño celular por sí misma, sino que aumenta el daño producido por otros mecanismos como la activación de la PKC y la glicosilación proteica6. Por otra parte, el papel de los polioles en el desarrollo de las complicaciones microvasculares de la diabetes ha sido constatado de manera indirecta mediante el empleo de inhibidores de la AR, como el sorbinil, el tolrestat y el ponalrestat7. Estas sustancias, que se han mostrado eficaces en la prevención de la retinopatía y la estabilización de la neuropatía, parecen disminuir el fenómeno de la hiperfiltración y reducir de forma leve la MAB. Sin embargo, el resultado global del uso estos compuestos ha sido algo desalentador, ya que algunos de ellos se han asociado con reacciones de hipersensibilidad y alteraciones de la función hepática, por lo que parece razonable pensar que por el momento no van a jugar un papel relevante en el tratamiento de la ERAD.
La proteína kinasa C
La PKC es una enzima de la familia de las serina-treonina kinasas que comprende quince isoformas que tienen en común el ser capaces de fosforilar las proteínas responsables de la transducción de señales intracelulares, y cuya consecuencia es la regulación de diversas funciones vasculares, que incluyen la contractilidad, el flujo, la proliferación celular y la permeabilidad vascular. La isoforma PKC-β2 aumenta su actividad en las células endoteliales de retina y riñón cuando éstas son expuestas a la hiperglucemia, debido al aumento en la síntesis de novo de DAG, un potente estimulador endógeno de esta enzima. Este aumento en la síntesis de DAG a partir de la hiperglucemia ocurre gracias a la activación de la vía de las pentosas y a una mayor oferta de dihidroxiacetonfosfato (DHAP). La PKC-β2, a su vez, activa la fosfolipasa A2, aumentando así la producción de prostaglandina PGE2 y de Tromboxano-A2. Estos últimos mediadores modifican drásticamente la permeabilidad endotelial y la respuesta a la Angiotensina II (AII) en el músculo liso vascular, cambios importantísimos en la génesis del daño renal presente en la diabetes.
Las células vasculares de los diabéticos muestran una activación de la PKC, cuyo papel se ha visto claramente implicado en el daño celular presente en la diabetes8. Así, el empleo de inhibidores de la PKC (LY-333531) y de la PKC-β2 se asocian a efectos beneficiosos a nivel renal, con reducción de la hiperfiltración glomerular, de la microalbuminuria (MAB), de la sobrexpresión del factor transformante del crecimiento-β (TGF-β) y del depósito de matriz extracelular9. Sin embargo, estudios clínicos recientes con ruboxistaurina en pacientes diabéticos con retinopatía no han mostrado ser eficaces en el control del daño renal10. Por otro lado, la variedad de isoformas de este grupo enzimático ha permitido inicar la búsqueda de nuevas dianas terapéuticas, como la PKC-α , que se ha propuesto como una posible alternativa ya que ratones knock-out para esta enzima no desarrollan MAB11.
Productos avanzados de la glicosilación
La glicosilación avanzada es el proceso no enzimático por el cual se produce la unión de azúcares reductores como la glucosa, a diferentes moléculas como proteínas. Este proceso tiene lugar en etapas sucesivas, las primeras son rápidas y reversibles, mientras que las finales son lentas e irreversibles: 1) La asociación del azúcar con la proteína, que resulta de la adición del grupo carbonilo del azúcar al grupo amino de la proteína, formando la denominada base de Schiff, estable por un corto tiempo. 2) Reordenamiento de los enlaces químicos, dando lugar a un producto más estable denominado genéricamente producto de Amadori. 3) El compuesto de Amadori sufre una serie de transformaciones que conducen a la formación de los AGEs. El mecanismo de estas reacciones no se conoce con detalle, aunque se sabe que involucran complejos reordenamientos intramoleculares y que, en algunos casos, la asociación entre varios de estos compuestos da lugar al entrecruzamiento entre distintas proteínas o entre distintas zonas de una misma proteína12.
Los AGEs se han relacionados con diferentes efectos a nivel renal, como la modificación de componentes estructurales de la membrana basal o de la matriz extracelular. Además, se han descrito receptores para estas moléculas (RAGE) que se expresan en diferentes localizaciones renales, incluyendo podocitos, células endoteliales y musculares lisas, células mesangiales y células epiteliales tubulares13. La unión a estos receptores determina la activación de diversas vías de señalización intracelular, con la subsiguiente generación de especies reactivas de oxígeno, la activación de factores de transcripción como el factor nuclear kappa B (NFkB), la liberación de citoquinas inflamatorias como el factor de necrosis tumoral-α (TNFα) o las interleukinas (IL) 1 y 6, la expresión de moléculas de adhesión y factores de crecimiento, como el TGF-β o el factor de crecimiento vascular endotelial (VEGF)13,14, elementos todos ellos que han sido relacionados con la patogenia del desarrollo y progresión de la ND. Además de trabajos in vitro y en modelos animales, hoy en día disponemos de estudios clínicos que implican de forma definitiva a los AGEs en el desarrollo de las complicaciones de la DM15,16.
Finalmente, la intervención sobre los AGEs como diana terapéutica ha demostrado potenciales beneficios. El contenido de AGEs en la dieta se ha sugerido como un factor de riesgo para el desarrollo de ND, habiéndose relacionado una dieta baja en AGEs con una reducción en las concentraciones de mediadores inflamatorios en pacientes diabéticos17, así como con un efecto nefroprotector en modelos animales18. Otra estrategia iría dirigida a la inhibición de la formación de AGEs. Así, estudios clínicos con el uso de pimagedina en pacientes diabéticos tipo 1 mostraron un enlentecimiento en la pérdida de filtrado glomerular, aunque sin beneficio significativo sobre la progresión de la nefropatía establecida19. Interesantemente, ha sido recientemente postulado que parte de los efectos renoprotectores no hemodinámicos derivados del uso de inhibidores de la enzima de conversión de la angiotensina (IECAs) pueden estar en relación con la inhibición de la formación de AGEs20. Otra aproximación se basa en el empleo de agentes que actúan rompiendo las uniones entre las moléculas que forman los AGEs (proteínas y azúcares). Estudios en modelos animales con este tipo de compuestos, como el ALT-711, han mostrado una atenuación del daño renal21,22. Finalmente, es posible inhibir la unión de los AGEs a sus receptores mediante el empleo de formas solubles de éstos o de anticuerpos neutralizantes dirigidos contra los RAGE, aproximaciones que han demostrado reducir la albuminuria y atenuar la expansión mesangial y la esclerosis glomerular en modelos animales23,24.
Estrés oxidativo
La alta actividad metabólica del riñón determina la generación de una importante cantidad de moléculas oxidantes, entre las que destacan las especies reactivas de oxígeno (ROS), como el peróxido de hidrógeno y el anión superóxido. Para su eliminación, el organismo dispone de un sistema de defensa antioxidante formado por elementos enzimáticos (superóxido dismutasa, glutatión peroxidasa, catalasa¿) y no enzimáticos (glutation, ácido ascórbico, α-tocoferol¿). El estrés oxidativo, situación en la que existe un exceso de estas moléculas altamente reactivas con capacidad oxidante, ha sido relacionado con importantes acciones deletéreas, como peroxidación lipídica, oxidación de proteínas, daño de ácidos nucleicos, inducción de factores de transcripción como NFkB, estimulación de la hipertrofia y proliferación celular, o inducción de apoptosis13,25.
La hiperglucemia es una situación inductora de estrés oxidativo, tanto a través de vías enzimáticas como no enzimáticas. Dentro de estas últimas se encuentran la auto-oxidación de la glucosa, los fenómenos de glicosilación avanzada, la vía de los polioles y, de manera crítica, las alteraciones del metabolismo mitocondrial, habiéndose sugerido que uno de los fenómenos iniciales en el desarrollo de las complicaciones de la DM es la formación de ROS por las mitocondrias26. En cuanto a las rutas enzimáticas, destaca la vía de la NADPH oxidasa, una importante ruta de producción de anión superóxido a nivel celular y vascular en pacientes diabéticos13. Todas las estructuras renales son susceptibles de sufrir daño oxidativo. En la ND ha sido demostrada la relación directa entre la severidad de la lesión renal y el grado de estrés oxidativo. Así, pacientes diabéticos con nefropatía establecida exhiben un mayor grado de daño oxidativo del ADN que pacientes con MAB, mientras que pacientes con un aumento en la excreción urinaria de albúmina (EUA) muestran mayores niveles de peroxidación lipídica que sujetos con normoalbuminuria28. Documentos histológicos demuestran la presencia de productos de glico-oxidación y lipo-oxidación en la matriz mesangial y en las lesiones nodulares glomerulares de pacientes diabéticos. Por otra parte, diversos estudios han observado que la actividad enzimática de superóxido dismutasa se encontraba reducida en sujetos diabéticos, indicando una asociación entre la alteración en la capacidad antioxidante y la ND29.
La intervención dirigida a reducir el estrés oxidativo ha sido postulada como una estrategia terapéutica de utilidad en la ND. Desde el punto de vista no farmacológico, la reducción de peso y la restricción de la ingesta de sodio han sido sugeridas como estrategias útiles30. En relación a las estrategias farmacológicas, se han empleado diversos antioxidantes para valorar su potencial beneficio como agentes renoprotectores, aunque los resultados, en general, no han sido consistentes13,25. El empleo de vitamina E, vitamina C o ácido lipoico en modelos animales demostró efectos beneficiosos sobre la reducción del estrés oxidativo y la protección de estructuras renales frente a este daño. Sin embargo, la traslación clínica de estos resultados ha sido limitada. Estudios clínicos sugieren que las vitaminas C y E pudieran mejorar la función endotelial en los pacientes diabéticos, aunque no hay datos concluyentes en relación a los beneficios sobre la ND. Lo que sí conocemos hoy en día es que los bloqueadores del sistema renina-angiotensina (SRA), así como las estatinas, fármacos que han sido relacionados con propiedades nefroprotectoras, asocian propiedades moduladoras sobre el estrés oxidativo. Así, estudios clínicos han demostrado que la adición de bloqueadores de los receptores AT1 de la angiotensina II a pacienes con ERC y proteinuria tratados con IECAs, resultó en un descenso de la peroxidación lipídica y de la oxidación de la albúmina urinaria31. En relación a las estatinas, el tratamiento de pacientes diabéticos tipo 2 hiperlipidémicos con simvastatina produjo, incluso antes de evidenciarse un descenso en los niveles de colesterol, una reducción en la generación de estrés oxidativo, indicando un efecto directo sobre este fenómeno32.
Factores de crecimiento y daño renal asociado a la diabetes
El papel de los fenómenos de inflamación y reparación tisular en la patogenia de las enfermedades renales se ha puesto de manifiesto en diversos estudios. En este sentido, las complicaciones microvasculares presentes en la diabetes constituyen un paradigma de la acción lesiva de numerosas citoquinas inflamatorias y factores de crecimiento33. Respecto a estos últimos, en los últimos años se ha observado la participación predominante de algunos de ellos, como son el factor de crecimiento transformante-β (TGF-β), el factor de crecimiento del endotelio vascular (VEGF) y el factor de crecimiento del tejido conectivo (CTGF) (Tabla 1).
Factor de crecimiento transformante-β
La superfamilia del TGF-β está formada por un gran grupo de polipéptidos extracelulares que participan en procesos de crecimiento, desarrollo, diferenciación y homeostasis a través de su interacción con receptores de la membrana celular. El TGF-β representa uno de los factores biológicos con mayor capacidad de generación de fibrosis, existiendo evidencias in vitro e in vivo de su acción inductora de la síntesis de procolágeno y colágeno, la generación de matriz extracelular-intersticial, así como de la inhibición de la degradación del colágeno por medio de la activación del PAI-1 (inhibidor del activador del plasminógeno). De manera esquemática, estos procesos se inician a partir de señales extracelulares que llegan al núcleo vehiculizadas por un tipo de proteínas fosforiladas conocidas como Smads (2 y 4), que van a producir la activación de la transcripción de determinados genes.
El TGF-β juega un papel patogénico significativo en diversas variedades de glomerulonefritis crónica, en la ERAD y en la nefropatía crónica del injerto34, actuando mediante la inducción y el mantenimiento de la fibrosis intersticial gracias a su efecto regulador sobre la proliferación celular, así como en la síntesis y degradación de la matriz extracelular. La hiperglucemia es un inductor de la producción de TGF-β. Así, en individuos sanos donde se mantiene una situación de hiperglucemia durante unos minutos se produce un aumento de la excreción urinaria de TGF-β y de isoprostanos. En el caso de la DM, es posible que la situación de hiperglucemia mantenida, a través de la activación de diferentes vías metabólicas junto con la hiperinsulinemia, sea uno de los factores inductores de la sobreexpresión de TGF-β, que como ya hemos indicado va a iniciar los fenómenos moleculares que producirán fibrosis intersticial y glomeruloesclerosis.
Estudios experimentales han constatado que el empleo de tiazolidíndionas (TZD) se asocia a una reducción de la sobreexpresión tisular de TGF-β junto a una reducción del acúmulo de pentosidina, observándose asimismo que la administración de insulina a cultivos de células tubulares aumentó la expresión del ARNm de TGF-β y la producción de la proteína. Este efecto abre nuevas perspectivas en la acciones renoprotectoras de las TZD e identifica tanto a la hiperinsulinemia como al TGF-β como posibles dianas terapéuticas, además de explicar parte de los mecanismos por los que estos fármacos son renoprotectores en pacientes obesos con ERAD35. Por otra parte, la administración de losartán en pacientes con nefropatía crónica del injerto es capaz de reducir los niveles de TGF-β, lo que sumado a la acción antiproteínurica de estos fármacos podría suponer un mecanismo adicional de renoprotección.
Factor de crecimiento del endotelio vascular
El VEGF es un potente mediador angiogénico endotelial con diferentes efectos biológicos, pero cuya función principal es la de mantener la integridad y viabilidad del endotelio. Su activación y acciones son fundamentales en numerosas situaciones, que incluyen los procesos de isquemia/reperfusión, inflamación, progresión de los fenómenos neoplásicos, situaciones de shock y reendotelización de superficies denudadas. El VEFG, que es producido por diferentes tipos celulares, entre los que se incluye la célula mesangial, actúa a través de la unión a receptores específicos de membrana con actividad tirosina quinasa (VEGFR1 y VEGFR2) y al receptor complementario neuropilina. La unión del VEGF a cada uno de sus receptores da lugar a la activación de varias vías de señalización, siendo el receptor VEGFR2 el más importante desde un punto de vista funcional. Varias citoquinas y factores de crecimiento pueden regular este gen, y a su vez, se han observado diferencias en su expresión cuando las células son sometidas a agentes agresores. Además, el control de la expresión del gen del VEGF ocurre tanto a nivel pre- como post-transcripcional. Los cambios en la tensión de oxígeno, a través de la inducción del factor inducido por hipoxia 1(HIF-1), son el mecanismo esencial en la regulación transcripcional del VEGF.
En el contexto de la DM, los diversos elementos que participan en la patogenia de esta enfermedad representan un estímulo para la producción de este factor, entre los que se encuentran los AGEs, el factor de crecimiento similar a la insulina (IGF), el TGF-β, la IL-1 y la IL-6. Sin embargo, el mecanismo es complejo, y en estudios realizados en cultivos de células tubulares y podocitos se ha observado que el medio hiperglucémico, aunque favorece la producción de VEGF, no es capaz de aumentar la angiogénesis. Recientemente se han constatado la participación del óxido nítrico (ON) junto al VEGF en los procesos de neovascularización presentes en la vasculopatía diabética36. El estudio de la expresión de ARNm de VEGF en biospias renales de pacientes con diferentes tipos de enfermedad renal ha demostrado que en las áreas de glomeruloesclerosis existe una disminución de las células que expresan VEGF, lo que se constata también en las células glomerulares en patologías como la amiloidosis, diabetes o el lupus eritematoso. Sin embargo, el daño en las células del epitelio visceral que se observa en varias de estas situaciones va a producir un aumento de la expresión de VEGF a nivel local, con el consiguiente aumento de la permeabilidad vascular y una alteración del funcionamiento de la célula endotelial37.
Las acciones terapéuticas relacionadas con este factor se dirigen a disminuir o aumentar las concentraciones tisulares de VEGF, con el objetivo de modificar el desarrollo de la vascularización. En general, estos tratamientos administran tanto el péptido de VEGF recombinante, para acción directa, como ADN u oligonucleótidos de VEGF para promover su síntesis. A nivel experimental se ha observado que el empleo de anticuerpos anti-VEGF en ratas diabéticas producía una disminución de la hiperfiltarción, de la albuminuria y de la hipertrofia glomerular38, previniendo asimismo la sobrexpresión de la ON sintasa endotelial (eNOS) en el capilar glomerular. Podría especularse sobre una posible relación patogénica entre la hiperglucemia y los cambios presentes en las fases precoces de la ERAD a través de esta vía, y la utilidad del VEGF como una posible diana terapéutica en fases iniciales de la enfermedad. Sin embargo, el hallazgo reciente de una disminución en la expresión del VEGF en el intersticio renal de pacientes con ERAD, a diferencia del aumento observado en algunos modelos animales, y su posible contribución a la progresión de la enfermedad39, ha despertado inquietud tanto sobre la validez de estos modelos para el estudio de algunos mecanismos fisiopatológicos donde interviene el VEGF como sobre la utilidad de su bloqueo como medida de prevención para el desarrollo de ERAD.
Factor de crecimiento del tejido conectivo
El CTGF es una citoquina que pertenece a una familia de proteínas modulares multifuncionales que actúan como moléculas asociadas a la matriz extracelular y regulan diversos procesos como la adhesión, migración, mitogénesis, diferenciación y supervivencia celular. En relación al CTGF, gran parte de su importancia radica en que muchos de los efectos del TGF-β, como por ejemplo la inducción de la síntesis de matriz extracelular, se encuentran mediados por la activación de este factor.
El CTGF se identificó por primera vez en células mesangiales expuestas a la hiperglucemia, y recientemente se ha demostrado su sobreexpresión en los podocitos, tanto en animales como en pacientes diabéticos. En estas situaciones, el CTGF favorece el daño glomerular a través de un aumento en la producción de proteínas de la matriz extracelular y de la inducción de cambios en la estructura del citoesqueleto40. Un reciente estudio en modelos murinos de diabetes tipo 1 y tipo 2 ha demostrado que la atenuación de la expresión del CTGF (knock-down) se asociaba a un menor grado de nefropatía, así como a una reducción de la expansión de la matriz mesangial y de los componentes que participan en la glomeruloesclerosis y la fibrosis intersticial41. Estos hallazgos indicarían que la mayoría de los efectos relacionados con el daño glomerular presente en la diabetes están mediados por el CTGF; sin embargo, los mecanismos son mucho más complejos. Así, recientemente se ha desarrollado un ratón transgénico que sobrexpresa el CTGF de manera exclusiva en los podocitos, a pesar de lo cuál no presenta anomalías glomerulares ni proteinuria. En cambio, en el modelo de diabetes por estreptozotocina esta sobrexpresión sí produce un aumento en la EUA y en el área mesangial, junto a vacuolización y reducción en el número de podocitos. Al comparar ambos modelos se observó en los animales transgénicos una menor actividad de la metaloproteinasa-2, enzima que participa en la degradación de la matriz mesangial.
El CTGF interviene como mediador en diversos procesos de daño renal mediante un efecto supresor sobre WNT y la proteína morfogénica ósea (BMP), y estimulador sobre TGF-β, receptores tipo integrina y receptores tipo neurotrofina. Sin embargo, todavía no se ha podido demostrar el papel de cada uno de ellos en la patogenia de la ERAD40, siendo algo prematuro proponer al CTGF como objetivo terapéutico en estas situaciones.
Sistema renina-angiotensina
El SRA es un determinante fundamental en los mecanismos que intervienen en el daño renal y vascular42. Además, este sistema controla la presión arterial (PA) y el balance hiodroelectrolítico a través de acciones coordinadas sobre el corazón, los vasos sanguíneos y los riñones. La AII, el principal efector del SRA, ejerce su efecto vasoconstrictor de manera predominante sobre las arteriolas eferentes del glomérulo, produciendo un aumento de la presión capilar glomerular y, como consecuencia, una mayor ultrafiltración de proteínas plasmáticas que contribuirá a la proteinuria, fenómeno importante en la aparición y progresión del daño renal. La AII también contribuye de manera directa a la progresión de la enfermedad renal mediante efectos no hemodinámicos, ya que actúa como como una verdadera citoquina favoreciendo el crecimiento celular, la inflamación y la fibrosis.
La síntesis y activación de los diferentes componentes del SRA se inicia con la producción de la renina en la aparato yuxtaglomerular (vía clásica), molécula que actúa sobre una proteína precursora circulante, el angiotensinógeno, para producir angiotensina I (AI). Este péptido tiene poco efecto sobre la PA, y a nivel de pulmonar se convierte en AII mediante la acción del enzima conversora de la angiotensina (ECA). La AII actúa en el corazón y los riñones uniéndose a la proteína G que se encuentra en los receptores tipo 1 (AT1) y tipo 2 (AT2). La activación del receptor AT1 pone en marcha los efectos deletéreos de la AII, como son la vasoconstricción y la hipertrofia vascular y cardiaca, junto a fenómenos inflamatorios, proliferativos y fibróticos. Una segunda forma de ECA (ECA-2) actúa sobre la AI produciendo la forma inactiva angiotensina 1-9, que a su vez, mediante la acción de la ECA, se convierte en angiotenisna 1-7, molécula con capacidad vasodilatadora y antiproliferativa, sobre todo en situaciones donde el SRA se encuentra más activado (ej. restricción de sodio). La ECA-2 está presente en el tejido renal humano, pero no se conoce con exactitud su distribución tisular en las situaciones de daño renal. En estudios en biopsias renales de pacientes con diferentes tipos de enfermedad renal se observó expresión de ECA-2 en el endotelio de los capilares glomerulares y peritubulares, y se pudo comprobar como el tratamiento con IECAs no modificaba su expresión43. La AII favorece la producción adrenal de aldosterona (Aldo), que recientemente ha sido reconocida como mediador en el daño renal y cardiaco. Al igual que la AII, también contribuye a la disfunción endotelial aumentando el tamaño y rigidez de las células endoteliales, lo que podría favorecer la pérdida de proteínas a través de las uniones intercelulares. Finalmente, se han descrito nuevas moléculas derivadas de la AII, como la AIV (corresponde a los péptidos 3-8), que se obtiene por la acción de angiopeptidasas, aminopeptidasas, carboxipeptidasas o endopeptidasas. Se conoce que la AIV se une de manera selectiva a su receptor (AT4) y favorece la secreción de PAI-1, aunque el papel biológico de estos nuevos péptidos es todavía incierto.
Numerosos estudios han demostrado que los fármacos que bloquean la actividad de los componentes del SRA ejercen un efecto renoprotector sobre la ERAD44,45. La capacidad renoprotectora de estos fármacos viene determinada tanto por su efecto sobre las acciones hemodinámicas de la AII, como sobre aquellas que se derivan de sus efectos proinflamatorios y profibróticos, efectos que se ejercen a través del estímulo directo sobre la síntesis de mediadores inflamatorios, como la IL-6, el factor activador de plaquetas (PAF) y los derivados del ácido araquidónico, y de factores de crecimiento como TGF-β, el factor de crecimiento derivado de las plaquetas (PDGF), el CTGF y el factor de crecimiento de fibroblastos (bFGF). Desde el punto de vista hemodinámico, uno de los principales objetivos para prevenir el desarrollo y progresión de la nefropatía, así como para reducir el riesgo cardiovascular en los enfermos diabéticos, es un estricto control de la PA (< 130/80 mmHg). En este contexto, los bloqueantes del SRA ofrecen una serie de beneficios terapéuticos diferenciales sobre el riesgo renal y cardiovascular no totalmente dependientes de su efecto sobre la PA, convirtiéndolos en fármacos antihipertensivos y antiproteinúricos de primera línea en el tratamiento de los enfermos con ND. Varios estudios han demostrado que el tratamiento con ARA es capaz de reducir la progresión de la lesión renal comparativamente con otros fármacos antihipertensivos para un mismo nivel de control tensional45. Para optimizar este efecto antiproteinúrico y renoprotector es necesario la titulación hasta alcanzar la dosis necesaria capaz de reducir la proteinuria o el empleo de la combinación de IECAs y ARA, cuyo efecto sobre la progresión de la ND frente a dosis adecuadas de cada uno de ellos por separado sigue siendo objeto de debate.
Por último, nuevos estudios han explorado el efecto sobre la progresión de la ERAD del bloqueo del SRA a otros niveles: inhibidores del la Aldo (espironolactona), bloqueadores del receptor de Aldo (eplerenona), inhibidores de vasopeptidasas y bloqueadores de la renina (aliskiren)42. La eplerenona se ha mostrado, tanto en monoterapia como en combinación con enalapril, eficaz en la reducción de la PA y la proteinuria en pacientes con DM tipo 2. El bloqueo del SRA en sus fases iniciales, evitando la reacción de la renina con el angiotensinógeno mediante Aliskiren, y su efecto sinérgico con otros fármacos que actúan sobre el SRA, constituye un área de interés en las opciones terapéuticas disponibles frente a la ERAD.
Inflamación y Nefropatía diabética
La relación entre inflamación y las complicaciones de la DM es un tema de gran interés, existiendo hoy en día evidencia sobre la estrecha interrelación entre inflamación y ND. Ha sido demostrada una asociación independiente entre proteína C reactiva (PCR) y EUA en pacientes diabéticos tipo 2. Del mismo modo, en este tipo de pacientes, estudios longitudinales han mostrado que el incremento de la EUA era significativa e independientemente determinado por los niveles de parámetros inflamatorios, sin existir diferencias en dicha asociación tras ajustar en función del índice de masa corporal, del aclaramiento de creatinina o de la presencia de HTA.
En los últimos años, diversos trabajos han señalado la importancia de las citoquinas inflamatorias como elementos determinantes del daño microvascular en la DM, y específicamente de la ND33,46 (Tabla 2). Las citoquinas son polipéptidos de bajo peso molecular que poseen acciones autocrinas, paracrinas y yuxtacrinas, con un importante papel inmunoregulador, pero también con significativos efectos pleiotrópicos. Estas moléculas son producidas tanto por células infiltrantes (principalmente macrófagos) como por las propias células renales (células endoteliales, mesangiales y tubulares).
Interleukina-1, Interleukina-6, Interleukina-18
Se ha demostrado un aumento en la expresión renal de IL-1 en modelos de ND, que se relaciona con el incremento en la expresión y síntesis de factores quimiotácticos y moléculas de adhesión a nivel de las células endoteliales y mesangiales47. Esta citoquina también se ha relacionado con anormalidades hemodinámicas intraglomerulares, disregulación en la síntesis de ácido hialurónico en las células epiteliales tubulares, así como con el aumento de la permeabilidad endotelial.
La IL-6 ha sido relacionada en la producción de alteraciones en la permeabilidad endotelial, la inducción de proliferación de las células mesangiales y el incremento de la expresión de fibronectina. Los niveles de expresión del ARN mensajero de IL-6 a nivel renal se relacionan de forma directa con la severidad del daño glomerular y de las alteraciones estructurales en la ND48. Asimismo, se ha observado un incremento en la expresión renal de IL-6 en modelos de ND, la cual se relaciona con índices de hipertrofia renal y con marcadores de daño glomerular47.
Respecto a la IL-18, se han objetivado niveles elevados de esta citoquina en pacientes con ND, mostrando una relación independiente con el incremento en la EUA y de β-2 microglobulina, así como con los cambios en la EUA durante la evolución de la nefropatía49. Más aún, un reciente estudio ha mostrado que los niveles séricos elevados de IL-18 pueden ser un predictor de disfunción renal en pacientes diabéticos con normoalbuminuria50.
Factor de Necrosis Tumoral-α
El TNFα es una molécula con actividades biológicas potencialmente implicadas en el daño renal del paciente diabético: efecto citotóxico directo sobre las células renales, inducción de apoptosis, alteración en la hemodinámica intrarenal, incremento en la permeabilidad endotelial o inducción de estrés oxidativo. Los niveles de expresión del ARN mensajero del TNFα y de la proteína se encuentran aumentados en el riñón de animales diabéticos47,51, relacionándose con las alteraciones iniciales en el desarrollo de la ND, como la hipertrofia renal y la hiperfiltración52,53. Además, los niveles de expresión y la excreción urinaria de TNFα se asocian de forma independiente con la EUA47,51, pudiendo observar un incremento significativo en los niveles de esta citoquina en orina y en el fluido intersticial renal precediendo al desarrollo de albuminuria. Finalmente, estudios clínicos han constatado que pacientes con ND muestran mayores concentraciones de esta citoquina que pacientes diabéticos sin datos de nefropatía, con una relación directa e independiente con marcadores de daño glomerular y túbulointersticial54.
Implicaciones terapéuticas
La creciente importancia de los mecanismos inflamatorios en el desarrollo y progresión de la ND señalan a este fenómeno como una diana terapéutica de potencial interés. Hoy sabemos que diversas estrategias que han mostrado efectos beneficiosos sobre la ND también presentan efectos moduladores sobre este fenómeno inflamatorio55. En este contexto, se ha comprobado que la retención de sodio y la hipertrofia renal observadas en las fases tempranas de la ND son prevenidas por el empleo de etanercept, una proteína dimérica de fusión producida genéticamente por tecnología de ADN recombinante, que interactúa con el receptor del TNFα humano-2 (FRNT2/p75) bloqueando la acción de esta citoquina. Del mismo modo, el empleo de infliximab, un anticuerpo monoclonal quimérico que neutraliza la actividad del TNFα, ha resultado en un descenso en la excreción urinaria de esta citoquina y una reducción de la EUA. Finalmente, la actuación sobre el TNFα es una estrategia que ha demostrado su aplicabilidad clínica en la ND basada en el uso de pentoxifilina, un fármaco que inhibe la transcripción del gen TNFα y reduce los niveles de ARN mensajero, modulando asimismo otras moléculas importantes dentro del proceso inflamatorio, incluyendo interferón-γ, IL-1β e IL-6.
La administración de pentoxifilina a pacientes diabéticos ha resultado en una reducción de marcadores clínicos de daño glomerular y túbulointersticial56,57. El efecto antiproteinúrico de este fármaco es similar al observado con el captopril58,59, y además, la administración conjunta de pentoxifilina y ARA o IECAs resulta en un efecto antiproteinúrico aditivo sobre el logrado por estos bloqueadores del SRA60,61.
Nefropatía diabética y Susceptibilidad genética
En el momento actual, no es posible predecir qué pacientes desarrollarán ND. Sabemos que solamente un porcentaje de los pacientes diabéticos va a desarrollar esta complicación, y que además, a pesar de una misma estrategia terapéutica, algunos presentarán una buena respuesta al tratamiento, mientras que otros permanecerán estables o progresarán hacia la insuficiencia renal. Todo ello sugiere la existencia de factores genéticos relacionados con el desarrollo y progresión de la ND, así como con la respuesta al tratamiento.
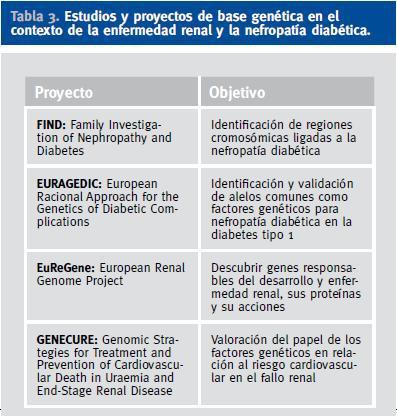
Actualmente existen diversos estudios y proyectos de base genética en el contexto de la enfermedad renal en general y de la ND en particular (Tabla 3)62. En la búsqueda de los genes responsables de la predisposición genética a la ND se han seguido dos estrategias de búsqueda principales: los estudios de genes candidatos y los rastreos del genoma. Los estudios de genes candidatos, o estudios de asociación, se basan en el análisis de genes que codifican proteínas que juegan un papel conocido en la fisiopatología de la enfermedad. Se trata de estudios de casos y controles donde se busca una posible asociación entre variantes alélicas de genes seleccionados a priori y el desarrollo de la enfermedad. Así, siguiendo esta estrategia, se ha observado que los pacientes portadores de la a-deleción en el intrón 4 del gen de la ON sintasa endotelial presentan una reducción en los metabolitos plasmáticos del ON, lo cual se ha postulado como un elemento facilitador del desarrollo de disfunción endotelial, y que se ha relacionado con un incremento del riesgo de ND avanzada63. Otros genes estudiados han sido los relacionados con el SRA, por ejemplo, un polimorfismo en el gen de la ECA basado en la inserción-deleción (I/D) de 287 pares de bases en el intrón 16, que condiciona que los individuos con genotipo DD presenten mayores niveles tisulares y circulantes de angiotensina II. Así, se ha postulado que los pacientes con el genotipo «desfavorable» (DD) tendrían más riesgo de desarrollar ND que el resto (ID/II), aunque la asociación entre ND y el genotipo DD en ambos tipos de diabetes es confusa y controvertida. Finalmente, un estudio muy reciente basado en la combinación de análisis de asociación casos-controles y de estudios funcionales ha sugerido que el alelo T del polimorfismo rs1617640 en la región promotora del gen de la eritropoyetina está significativamente asociado con la insuficiencia renal terminal en pacientes diabéticos64.
La otra estrategia de búsqueda son los estudios de ¿rastreo del genoma¿. Se basan en la búsqueda en familiares de la presencia de ligamiento genético para heredar determinadas regiones del genoma y el desarrollo de la enfermedad. Actualmente, una de las iniciativas más importantes que emplea esta estrategia es el ¿Family Investigation of Nephropathy and Diabetes (FIND) Study¿65, un consorcio multicéntrico constituido con el objetivo de identificar genes responsables del desarrollo de ND por el Instituto Nacional de Diabetes y Enfermedades Digestivas y Renales de Estados Unidos. Hasta el momento, la evidencia más robusta de ligamiento en relación a la ND señala a regiones en los cromosomas 7 (7q21.3), 10 (10p15.3), 14 (14q23.1) y 18 (18q22.3), confirmando resultados previos en relación a regiones de ligamiento en los cromosomas 7, 10 y 18, lo cual indica la relevancia de estudiar los genes contenidos en estas regiones cromosómicas en relación a la identificación de genes de susceptibilidad para la ND.
En conclusión, la existencia y persistencia en la DM de un ambiente hiperglucémico, junto al efecto que éste ejerce sobre gran variedad de factores (sistemas enzimáticos, citoquinas, factores de crecimiento, etc), así como las interacciones entre ellos, va a condicionar el desarrollo y la progresión de la ERAD. Este complejo entramado de factores e interacciones está matizado por factores genéticos que pueden modular en uno u otro sentido la evolución de la enfermedad y la respuesta a las diferentes posibilidades de tratamiento. Todo ello nos obliga a establecer estrategias terapéuticas complejas y a mantener el necesario equilibrio riesgo/beneficio en aras de conseguir una respuesta óptima, con las máximas garantías, y preservando los criterios de eficiencia y evidencia científica.
PUNTOSCLAVE
1. La hiperglucemia es el factor determinante en el desarrollo de las complicaciones asociadas a la diabetes, y su control es fundamental desde un punto de vista preventivo.
2. La hiperglucemia y el incremento de la glucosa intracelular resultan en la activación de vías metabólicas alternativas, como la vía de los polioles, con la participación determinante de elementos enzimáticos como la aldosa-reductasa. La inhibición de la actividad de esta enzima ha demostrado su utilidad en la retinopatía y neuropatía diabéticas, pero los efectos potencialmente beneficiosos sobre la nefropatía no han encontrado una adecuada traslación clínica por el momento.
3. A pesar de que la protein kinasa C es un elemento clave en la génesis del daño celular en la diabetes, y de que en modelos animales su inhibición se asocia a efectos beneficiosos a nivel renal, estudios clínicos no han demostrado de forma consistente la eficacia de esta estrategia en el tratamiento de la nefropatía diabética.
4. Los productos avanzados de la glicosilación han sido relacionados con la patogenia y progresión de la nefropatía diabética. Existen diversas aproximaciones terapéuticas basadas en la inhibición del efecto de estos productos, desde reducir su contenido en la dieta hasta bloquear la unión a sus receptores. Dichas aproximaciones han demostrado su utilidad en modelos animales y en algunos estudios clínicos donde se ha objetivado el enlentecimiento de la pérdida de filtrado glomerular en pacientes afectos de diabetes tipo 1.
5. En la nefropatía diabética ha sido demostrada la relación directa entre la severidad de la lesión renal y el grado de estrés oxidativo. A pesar de ello, y aunque hay datos indirectos del posible beneficio de la reducción de este fenómeno, el empleo de antioxidantes no ha demostrado un claro efecto renoprotector.
6. Los factores de crecimiento están involucrados en la fisiopatología de la nefropatía diabética, con la participación predominante de algunos de ellos, como el factor de crecimiento transformante-β, el factor de crecimiento del endotelio vascular y el factor de crecimiento del tejido conectivo. E stas moléculas son potenciales dianas terapéuticas que están bajo intensa investigación en el momento actual.
7. Ha sido demostrado con evidencia sólida el papel crítico del sistema reninaangiotensina-aldosterona en el desarrollo y progresión de la nefropatía diabética. La inhibición de este sistema a sus distintos niveles es, hoy en día, una de las estrategias de tratamiento más importantes en estos pacientes, y continúa siendo un área de investigación de gran interés.
8. Una de las más recientes y novedosas aportaciones al conocimiento de la fisiopatología de la nefropatía diabética deriva de la participación del fenómeno inflamatorio, más específicamente de las citoquinas inflamatorias, lo que abre nuevas vías de investigación y tratamiento.
9. Vamos conociendo cada vez con mayor detalle cómo la variabilidad genética entre los distintos individuos es un elemento determinante en la diferente susceptibilidad al desarrollo y progresión de la nefropatía diabética, y como es asimismo un factor importante que dertermina la respuesta a las diversas estrategias terapéuticas.

Figura 1.

Tabla 1. Acciones biológicas de los factores de crecimiento involucrados en la fisiopatología de la nefropatía diabética

Tabla 2. Acciones biológicas de las citoquinas inflamatorias potencialmente involucradas en la fisiopatología de la nefropatía diabética

Tabla 3. Estudios y proyectos de base genética en el contexto de la enfermedad renal y la nefropatía diabética


