
La reabsorción tubular de calcio es uno de los principales factores que determinan la concentración sérica de calcio y su excreción urinaria. El túbulo contorneado distal y conector es donde se produce la regulación fina de la calciuria. A ese nivel se encuentra el canal epitelial de Ca (TRPV5), que es el paso limitante en el transporte transcelular de Ca. La presencia dinámica del canal TRPV5 en la superficie de la célula tubular está mediada por un proceso de reciclado endosómico. Distintos factores intrarrenales intervienen en la fijación del canal de calcio en la membrana aplical, entre ellos la hormona antienvejecimiento klotho y la calicreína tisular (CT). Ambas proteínas son sintetizadas en el túbulo distal y secretadas en el fluido tubular. La calicreína tisular estimula la reabsorción activa de calcio por vía del receptor de bradiquinina tipo 2 que compromete la activación del of TRPV5 por vía de la protein cinasa C. Los ratones deficientes en CT muestran hipercalciuria de origen renal comparable a la pérdida de calcio que se observa en los ratones knockout para el TRPV5. Existe un polimorfismo con pérdida de función del gen de la CT humana denominado R53H (alelo H) que produce una gran disminución de la actividad enzimática. La presencia del alelo H, por lo menos en la población japonesa, parece ser frecuente (24%). Estos individuos tiene una tendencia a excreción más alta de calcio y sodio en orina que se manifiesta más durante la infusión de furosemida. En el futuro habrá que estudiar si la manipulación del sistema calicreína-quinina renal permite corregir la hipercalciuria idiopática con fármacos diferentes a los diuréticos tiazídicos.
Renal tubular calcium reabsorption is one of the principal factors that determine serum calcium concentration and calcium excretion. Calcium excretion is regulated by the distal convoluted tubule and connecting tubule, where the epithelial calcium channel TRPV5 can be found, which limits the rate of transcellular calcium transport. The dynamic presence of the TRPV5 channel on the surface of the tubular cell is mediated by an endosomal recycling process. Different intrarenal factors are involved in calcium channel fixation in the apical membrane, including the anti-ageing hormone klotho and tissue kallikrein (TK). Both proteins are synthesised in the distal tubule and secreted in the tubular fluid. TK stimulates active calcium reabsorption through the bradykinin receptor B2 that compromises TRPV5 activation through the protein kinase C pathway. TK-deficient mice show hypercalciuria of renal origin comparable to that seen in TRPV5 knockout mice. There is a polymorphism with loss of function of the human TK gene R53H (allele H) that causes a marked decrease in enzymatic activity. The presence of the allele H seems to be common at least in the Japanese population (24%). These individuals have a tendency to greater calcium and sodium excretion in urine that is more evident during furosemide infusion. Future studies should analyse if manipulating the renal kallikrein-kinin system can correct idiopathic hypercalciuria with drugs other than thiazide diuretics.
La reabsorción tubular de calcio es uno de los principales factores que determinan la concentración sérica de calcio y su excreción urinaria. La mayor parte del Ca filtrado (60-70%) es reabsorbido en el túbulo proximal, primariamente por un mecanismo paracelular que no es sensible de una forma relevante a las hormonas reguladoras del Ca. Otro 20-25% del Ca filtrado es reabsorbido en la rama gruesa ascendente de Henle primariamente por vía paracelular, e involucra las claudinas 16, 19 y 14. El túbulo contorneado distal y conector es donde se produce la regulación fina de la calciuria, y donde se reabsorbe una fracción significativa (10-15%) de la carga filtrada de Ca. En este segmento tubular el Ca se reabsorbe por un mecanismo transcelular, entrando a través de canales de Ca presentes en la membrana apical. Bajo condiciones normales, la reabsorción tubular de Ca está estrechamente regulada. Factores no hormonales, como el volumen del LEC, el estado ácido/base y las concentraciones plasmáticas de magnesio y Ca ejercen una influencia sobre el manejo del Ca en el túbulo renal1–3. Factores hormonales extrarrenales, como la hormona paratiroidea y la 1,25-dihidroxivitamina D, también regulan la reabsorción tubular de Ca4. En contraste, relativamente poco se conoce sobre la posible contribución de factores intrarrenales en la regulación del transporte tubular renal de Ca.
Canal epitelial de calcio TRPV5 y su regulación por factores tubularesRecientemente se ha descubierto la base molecular del transporte activo transcelular de Ca en el nefron distal. Este proceso involucra el influjo apical de Ca a través del canal epitelial de Ca (TRPV5), que es el paso limitante en el transporte transcelular de Ca5. Consistente con ello, la falta de canal TRPV5 lleva a una disminución en la reclamación tubular distal de Ca y la producción de hipercalciuria de origen renal6. Varias hormonas calciotrópicas, conocidas por alterar la reabsorción renal de Ca, afectan la expresión del TRPV5. Otras estimulan el tráfico del canal TRPV5 hacia la membrana plasmática, mientras que cierto número de iones y proteínas asociadas controlan la actividad del canal a nivel de la membrana plasmática7. La presencia dinámica del canal TRPV5 en la superficie de la célula tubular es mediada por un proceso de reciclado endosómico que permite internalizar el canal para hacerlo reaparecer nuevamente a nivel de la membrana plasmática. Una de las proteínas sintetizadas por el túbulo distal es la hormona antienvejecimiento klotho. Klotho es una proteína transmembrana de un solo paso, expresada principalmente en los riñones y en el plexo coroideo. El klotho de membrana funciona como un correceptor obligado del factor de crecimiento fibroblástico 23 (FGF-23) en el riñón y la glándula paratiroidea. El dominio extracelular de klotho está compuesto de 2 repeticiones internas, KL1 y KL2, que pueden ser clivadas y liberadas hacia la sangre y hacia la luz tubular y actuar como hormonas. Klotho up regula el TRPV5 tanto desde dentro como desde fuera de la célula tubular. La acción intracelular del klotho es probable que sea debida a un aumento de tráfico de proteínas del canal hacia la membrana apical, mientras que su acción extracelular sería debida a la inhibición de la endocitosis de las caveolas donde se encuentran los canales de Ca. Ambos efectos comprometen la actividad de sialidaza del klotho, modificando el estado de glucosilacion del canal de Ca y, por lo tanto, atrapando el canal a nivel de la superficie celular8. Consistente con la influencia positiva del klotho sobre la reabsorción de Ca mediada por el TRPV5 es que los ratones a los cuales les falta el gen klotho tienen pérdida urinaria de Ca, hiperparatiroidismo e hipervitaminosis D9.
Otra de las proteínas que son sintetizadas en el túbulo distal y secretadas en el fluido tubular es la calicreína tisular (CT)10–12. La CT es una serino-proteasa comprometida en la generación de quininas en muchos órganos, incluido el riñón13. Las quininas generadas en el túbulo colector a través de la CT inhibe la reabsorción de ClNa, a través de la activación de los receptores B2 de la bradiquinina localizados a lo largo de las células epiteliales del túbulo colector. Estas quininas son inmediatamente inactivadas por 2 enzimas riñón-específicas (quininasas), la exopeptidasa carboxipeptidasa Y-like (CPY), y la endopeptidasa neutra (NEP)14 (fig. 1). Por otro lado, la CT, a través de su actividad catalítica, actúa directamente sobre el canal epitelial de sodio (ENaC) con el objeto de modular su actividad, sin ser crítica para la regulación renal de la homeostasis del sodio. Los ratones deficientes en CT exhiben una absorción neta transepitelial del K+ en el túbulo cortical colector debido a que se produce una activación anormal de H+, K+- ATPasa tipo colónica en las células intercalares y una reducida secreción de K+ por las células principales, secundaria a una disminución de la actividad del ENaC. Por lo tanto, la CT es un factor caliurético que provee una protección rápida e independiente de la aldosterona contra la hipercalemia, luego de una carga dietaria de K15. La deficiencia parcial de CT en humanos impide una adecuada adaptación a una carga de potasio16. Recientemente se ha demostrado que los ratones deficientes en CT muestran hipercalciuria de origen renal17 comparable al leak de Ca que se observa en los ratones knockout para el TRPV5. La distribución de la CT se superpone principalmente con aquella del canal TRPV5 en el nefrón distal, y la expresión del gen de calicreína se ve incrementada en las dietas bajas en Ca. Recientemente se ha demostrado que la CT estimula la reabsorción activa de Ca por vía del receptor de bradiquinina tipo 2 que compromete la activación del TRPV5 por vía de la protein cinasa C18. Esto indica que la calicreína podría ser un modulador fisiológico de la reabsorción tubular de Ca.
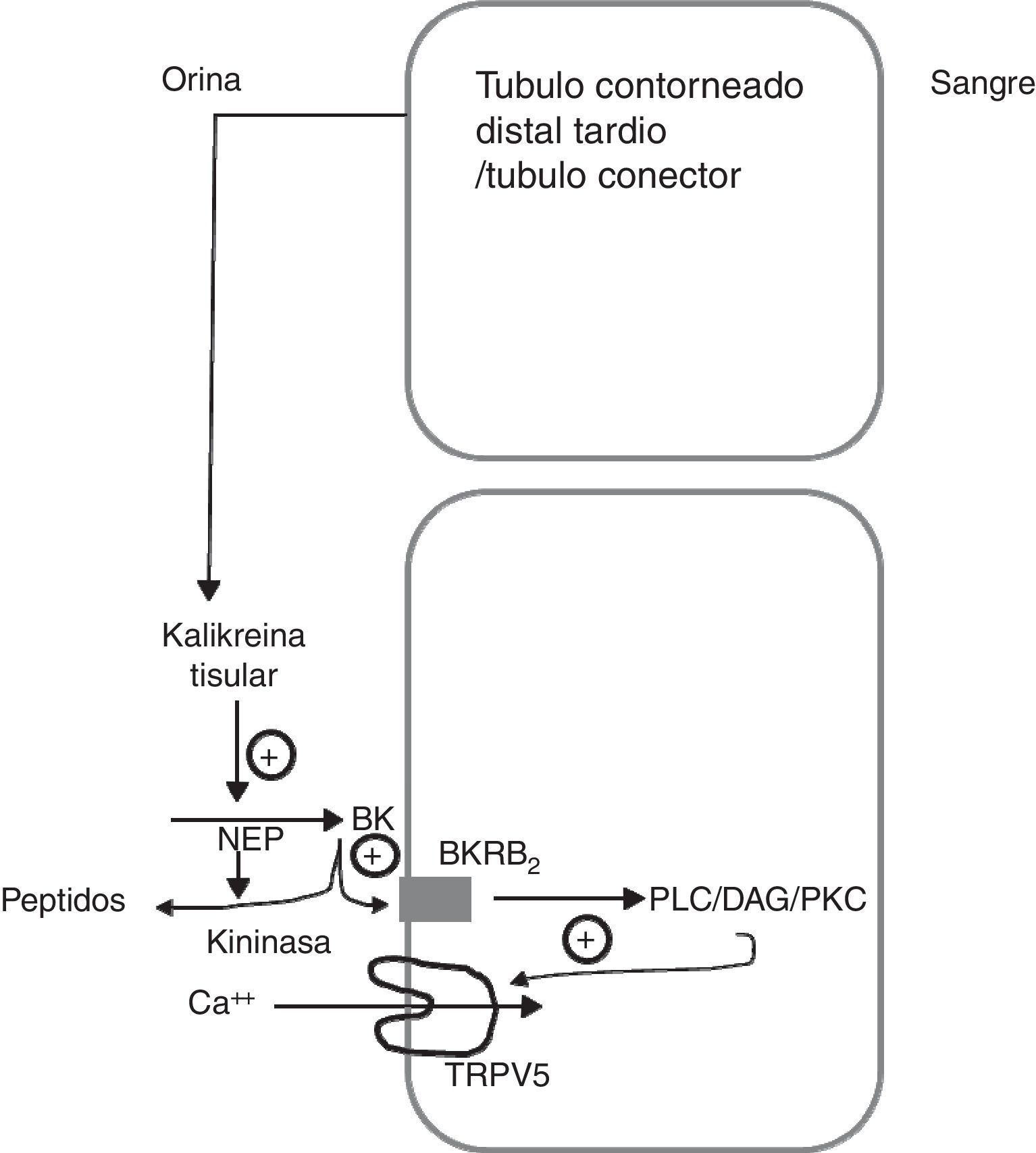
 filtrado o segregado localmente y produce finalmente bradiquinina (BK). La BK actúa sobre su receptor B2 (BKRB2) activando la vía de fosfolipasa C/diacilglicerol/proteincinasa C (PLC/DAG/PKC) induciendo la localización del canal de calcio TRPV5 a nivel de la membrana apical y favorece la reabsorción de calcio tubular. La BK es degradada por la endopeptidasa neutra (NEP) y la quininasa CYP renal.")
Modelo esquemático que muestra como la calicreína tisular participa de la regulación del canal epitelial de calcio TRPV5 en el túbulo contorneado distal tardío.
La calicreína tisular producida por el túbulo conector es liberada dentro del fluido urinario. Alli actua sobre el quininogeno (QN) filtrado o segregado localmente y produce finalmente bradiquinina (BK). La BK actúa sobre su receptor B2 (BKRB2) activando la vía de fosfolipasa C/diacilglicerol/proteincinasa C (PLC/DAG/PKC) induciendo la localización del canal de calcio TRPV5 a nivel de la membrana apical y favorece la reabsorción de calcio tubular. La BK es degradada por la endopeptidasa neutra (NEP) y la quininasa CYP renal.
La secreción de calicreínas renales por las células del túbulo colector puede ser incrementada por los bloqueadores de canales de potasio ATP dependientes (KATP). Así, la administración oral de glibenclamida (un bloqueante de los canales KATP) o de un bloqueante de los canales KATP riñón-selectivo, el U18177, induce natriuresis y un incremento en secreción de calicreínas que suprimen la elevación de la presión sistólica en ratas Sprague Dawley que recibieron ClNa al 8% en su dieta. Estos efectos se ven anulados con la coadministración de un antagonista del receptor B2 de bradiquinina (B2BK), FR17365, que indica que el bloqueo de los canales KATP induce la diuresis y natriuresis por la liberación de calicreínas renales19.
La ebelactona B y la poststatina son inhibidores de la quininasa renal CPY. Cuando estos fármacos se usan en un modelo de hipertensión inducida por sal-DOCA, previenen el desarrollo de hipertensión sal dependiente19. Un inhibidor de la NEP, BP102, también suprime la elevación de la presión arterial sistólica, pero su efecto es más débil con respecto a la ebelactona B. Curiosamente, las ratas SHR jóvenes genéticamente hipertensas que presentan hipercalciuria tienen una capacidad atenuada para secretar calicreínas renales en comparación con las ratas normotensas WKY20. Hasta el momento no hay estudios que hayan manipulado el sistema calicreína-quinina renal con los fármacos anteriormente mencionados para ver si modifican la calciuria en animales.
Deficiencia de calicreína tisular en humanos y su efecto sobre la calciuriaExiste un polimorfismo con pérdida de función del gen de la CT humana denominado R53H (alelo H) que produce una gran disminución de la actividad enzimática. En un estudio cruzado en individuos jóvenes blancos varones, 30 de los sujetos eran homocigotos para el R53H y 10 eran heterocigotos para el mismo gen. Fueron asignados al azar a periodos de 7 días de dieta baja en Ca (10mmol/día) asociado con una dieta baja en Na/alta en K o una dieta alta-Na/baja-K para modular la síntesis de CT21. Al séptimo día de cada dieta, los participantes fueron estudiados antes y durante la infusión de 2 h de furosemida, que funcionalmente excluye la rama gruesa ascendente de Henle e incrementa la oferta de Ca a los segmentos tubulares distales. La actividad de calicreína urinaria fue un 50-60% más baja en los participantes con R53H que en aquellos sin el R53R. La adaptación de la excreción urinaria de Ca a las dietas contrastantes en Na/K no fue afectada en los participantes portadores de R53H. En contraste, los participantes R53H luego de la infusión de furosemida tuvieron significativamente concentraciones de Ca ionizado sérico más bajas que los participantes R53R (p<0,0001) y una tendencia no significativa a mayores excreciones urinarias de Ca que los participantes R53R. Estos efectos fueron más marcados con dietas bajo-Na/alto-K.
Otro estudio reciente analizó polimorfismos de la CT en voluntarios japoneses y examinó la asociación entre el alelo H en la región del promotor, que como ya dijimos ha mostrado producir una disminución de la actividad del promotor, y la actividad de la calicreína urinaria22. Fueron analizados para el promotor y las regiones codificantes del gen de la calicreína tisular 90 y 73 voluntarios, respectivamente. La presencia del alelo H en la población japonesa fue frecuente, del 24%. Se encontró un polimorfismo sinónimo y 3 no sinónimos en las regiones codificantes. Estudiaron los parámetros fisiológicos en sujetos tratados mediante una dieta ad libitum. La actividad de calicreína urinaria no estuvo disminuida significativamente en los sujetos con el alelo H comparados con los individuos sin el alelo H, a pesar de que estuvo baja en 2 homocigotos para el alelo H. Sin embargo, la excreción urinaria de Ca y Na fueron mayores en los sujetos con el alelo H aquellos sin ese alelo.
De estos estudios se deduce que el polimorfismo con pérdida de función del gen de la CT humana (alelo H) es relativamente frecuente. Estos individuos tiene una tendencia a excreción más alta de Ca y Na en orina y a que el defecto se manifieste más durante la infusión de furosemida, que altera la reabsorción de Ca en esa porción tubular, exagerando los defectos de reabsorción en el túbulo distal.
ColofónEn el futuro habrá que estudiar si el sistema calicreína-quinina renal está alterado en animales con hipercalciuria y en humanos con hipercalciuria idiopática. Este sistema podría ser modificado por bloqueantes de los canales KATP riñón-selectivos que aumentan la secreción de calicreína tisular renal o a través del aumento de la quininas urinarias por inhibidores de la quininasa renal CPY, como la ebelactona B y la posestatina. De esta manera se podría tratar la litiasis renal recurrente producida por hipercalciuria idiopática por fármacos diferentes a los diuréticos tiazídicos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



