
Renal diseases associated with hypomagnesemia are a complex and diverse group of tubulopathies caused by mutations in genes encoding proteins that are expressed in the thick ascending limb of the loop of Henle and in the distal convoluted tubule. In this paper, we review the initial description, the clinical expressiveness and etiology of four of the first hypomagnesemic tubulopathies described: type 3 Bartter and Gitelman diseases, Autosomal recessive hypomagnesemia with secondary hypocalcemia and Familial hypomagnesemia with hypercalciuria and nephrocalcinosis. The basic biochemical patterns observed in renal tubular hypomagnesemias and the modalities of transport and interaction that occur between the transporters involved in the reabsorption of magnesium in the distal convoluted tubule are described below. Finally, the recent report of a new renal disease with hypomagnesemia, type 2 hypomagnesemia with secondary hypocalcemia caused by reduced TRPM7 channel activity is described.
Las enfermedades renales que cursan con hipomagnesemia son un grupo complejo y variopinto de tubulopatías producidas por mutaciones en genes que codifican proteínas que se expresan en la rama gruesa ascendente del asa de Henle y en el túbulo contorneado distal. En el presente artículo revisamos la descripción inicial, la expresividad clínica y la etiología de cuatro de las primeras causas de tubulopatías hipomagnesémicas que se describieron: las enfermedades de Bartter tipo 3 y Gitelman, la hipomagnesemia con hipocalcemia secundaria autosómica recesiva y la hipomagnesemia familiar con hipercalciuria y nefrocalcinosis. A continuación, se describen los patrones bioquímicos básicos que se observan en las hipomagnesemias tubulares renales y las modalidades de transporte e interacción que concurren entre los transportadores implicados en la reabsorción de magnesio en el túbulo contorneado distal. Finalmente, se comunica la reciente descripción de una nueva tubulopatía hipomagnesémica, la hipomagnesemia con hipocalcemia secundaria tipo 2 causada por una reducción de la actividad del canal TRPM7.
Renal tubules select the filtered solutes necessary to maintain body homeostasis. Most solutes are reabsorbed and returned to the blood; those that are not needed will be eliminated in the urine.
In the first tubular regions the process of reabsorption is very intense (for example, 65% of the sodium filtered is reabsorbed in the proximal tubule and 25% in the ascending branch of the loop of Henle). For this reason, tubulopathies with intense damage in these segments (Toni-Debré-Fanconi syndrome and Bartter syndrome, for example) can be very serious and difficult to treat due to the difficulty of compensating for the loss of solutes.
It is therefore up to the distal tubule and collecting duct to fine-tune, to put the finishing touches on recovering the solutes necessary for the body and therefore must not be lost in the urine. This task is very complex and requires appropriate specialization.
Distal convoluted tubuleTo highlight its importance, it is enough to recall the functional interest of the macula densa as a sensor of intratubular sodium concentration within the structure of the juxtaglomerular apparatus. Suffice to emphasize that the distal tubule contains the only renal specific channel for calcium reabsorption (ECaC) whose function is modulated by various hormones and that the regulation of the thiazide-sensitive sodium chloride (NaCl) transporter is very complex, as can be seen when studying the pathophysiology of Gordon’s syndrome. In addition, the distal tubule has an important functional involvement in calcium and magnesium reabsorption (Mg2+) and has the highest Na+, K+-ATPase activity of the nephron in the basolateral membrane and supplies the energy necessary for ionic transport in this segment. The complexity of the regulation of tubular Mg reabsorption2+ is manifested in the large number and diversity of hypomagnesemic tubulopathies.
Physiology of renal tubular handling of Mg2+The ion Mg2+ is filtered by the glomerulus. In the proximal tubule, 10%–25% of the initially filtered load is reabsorbed via a passive paracellular pathway that is poorly understood. The ascending thick branch of the loop of Henle reabsorbs approximately 50%–70% of the filtered Mg load2+ via passive paracellular transport. The remaining 5%–10% of the filtered Mg2+ is reabsorbed in the distal convoluted tubule via an active transcellular route. Thus, the causes of renal hypomagnesemia are in the loop of Henle and distal tubule. In the end, more than 96% of the filtered Mg2+ is reabsorbed along the nephron.
Historical description of the main causes of hypomagnesemia- a)
In 1962, Bartter et al. reported clinical and biochemical data observed in two male patients aged 5 and 25 years respectively, with a new disease characterized by growth retardation, hyperplasia of the juxtaglomerular apparatus, hyperaldosteronism, normal blood pressure, hypokalemic and hypochloremic metabolic alkalosis, and defect in renal concentrating ability resistant to the action of pitresin.1 It eventually became clear that the term Bartter syndrome refers to up to five diseases that share a defective reabsorption of salt, potassium, calcium and water in the ascending limb of the loop of Henle. Two clinical patterns were established that allow distinguishing between a severe form of antenatal presentation (neonatal Bartter corresponding to types 1 and 2) and a form of later onset during the first years of life (classic Bartter or type 3).
- b)
In 1966, Gitelman et al. published clinical data on three adult patients, two of them brothers, with hypokalemia, hypomagnesemia and metabolic alkalosis.2 For many years, patients with these features were misdiagnosed as a variant of Bartter syndrome. The presence of hyperreninemia and hyperaldosteronism contributed to this confusion. Clinical onset usually appears in adolescence, generally with mild neuromuscular symptoms. The spectrum of manifestations, however, is broad. Thus, the disease may be asymptomatic or manifest itself as mild and sometimes intermittent symptoms (muscle weakness, cramps, fatigue, polyuria, nocturia or joint pain) or more severe symptoms (tetany, convulsions). Paresthesias occur frequently, especially in the face. Salt craving is common and blood pressure values are lower than in the general population. Growth is usually unaffected, although failure to thrive and short stature have been reported in a minority of cases. Hypomagnesemia and hypokalemia prolong ventricular repolarization which predisposes to severe arrhythmias.
- c)
In 1968, Paunier et al. reported the case of a male infant who at six weeks of age presented with generalized seizures and tetany associated with hypomagnesemia and hypocalcemia.3 The hypomagnesemia was associated with elevated fractional excretion of Mg2+. Treatment with Mg2+ corrected tetany and normalized calcium levels. Renal biopsy showed hyalinization of some glomeruli, interstitial fibrosis and calcium salt deposition. This was the first case of what later became known as hypomagnesemia with secondary hypocalcemia. Shortly thereafter, another case was published in a five-month-old infant with tetany due to secondary hypomagnesemia and hypocalcemia.4 Treatment with vitamin D corrected the hypocalcemia without changing the clinical situation. The child died shortly thereafter; biopsy showed calcinosis of the myocardium, kidneys and in one of the cerebral arteries.4 Later, in another family, the association with incomplete distal renal tubular acidosis was described.5 In the cases described later, muscle cramps and convulsions coincided.
- d)
Although some cases combining hypomagnesemia and nephrocalcinosis had been reported6–8 previously, the first article reporting the presence of hypomagnesemia and nephrocalcinosis was related to hypercalciuria and bilateral macular coloboma appeared in 1979.9 The authors reported that the condition suffered by the two affected siblings could be included in a new variant of the oculorenal syndrome because it had some similarities with Lowe syndrome.9
Subsequently, some cases of isolated tubular hypomagnesemia of unknown origin were published. Thus, hypomagnesemias of renal tubular origin were not systematized well into the 1980s. The Spanish pediatric nephrologist Juan Rodríguez Soriano and his collaborators described in 1987 in an anthological article that at least three hereditary diseases with hypomagnesemia should be considered, caused by defects in renal tubular reabsorption of Mg2+, namely, familial isolated hypomagnesemia, familial hypomagnesemia–hypokalemia or Gitelman syndrome and familial hypomagnesemia–hypercalciuria.10 Of the latter, at least 15 patients had been described, to which the authors added three new cases. The authors wrote that “Hypomagnesaemia is always accompanied by hypercalciuria and nephrocalcinosis. Ocular abnormalities such as myopia and horizontal nystagmus are often present. Hypermagnesiuria is of a greater degree than that observed in the previous entity and reflects a low [maximal transport] of Mg reabsorption.” They further emphasized that, “The defect must be situated at the level of the ascending limb of the loop of Henle and affects the transport of both calcium and Mg2+”.10
In 1995, Praga et al., members of the Hospital 12 de Octubre in Madrid, published clinical and biochemical data corresponding to eight patients belonging to five families with familial hypomagnesemia–hypercalciuria. The authors found that, in this autosomal recessive tubulopathy, there is a progressive deterioration of renal glomerular function and that the tubular damage did not recur after transplantation. The title of their article is the one that has passed to posterity and given the disease its name: familial hypomagnesemia with hypercalciuria and nephrocalcinosis.11
Molecular genetics describes the etiology of the four tubulopathies mentioned aboveSince the mid-1990s and within seven years, the etiology of the four causes of hypomagnesemia mentioned above was described. Later, other variants were discovered and have recently been reviewed by our group.12
- a)
Bartter’s syndrome type 3 is caused by recessive mutations that result in a loss of function of the CLCNKB gene encoding a Cl channel− (ClC-Kb) that is expressed in the basolateral membrane of the ascending thick branch cells of the loop of Henle and in the distal convoluted tubule13 (Fig. 1). Loss of ClC-Kb function alters the intracellular regulation of Cl- concentration which subsequently interferes with the generation of the lumen-positive potential resulting in salt loss and possibly hypomagnesemia. Patients with this subtype develop reduced Mg levels2+ during infancy or thereafter.14
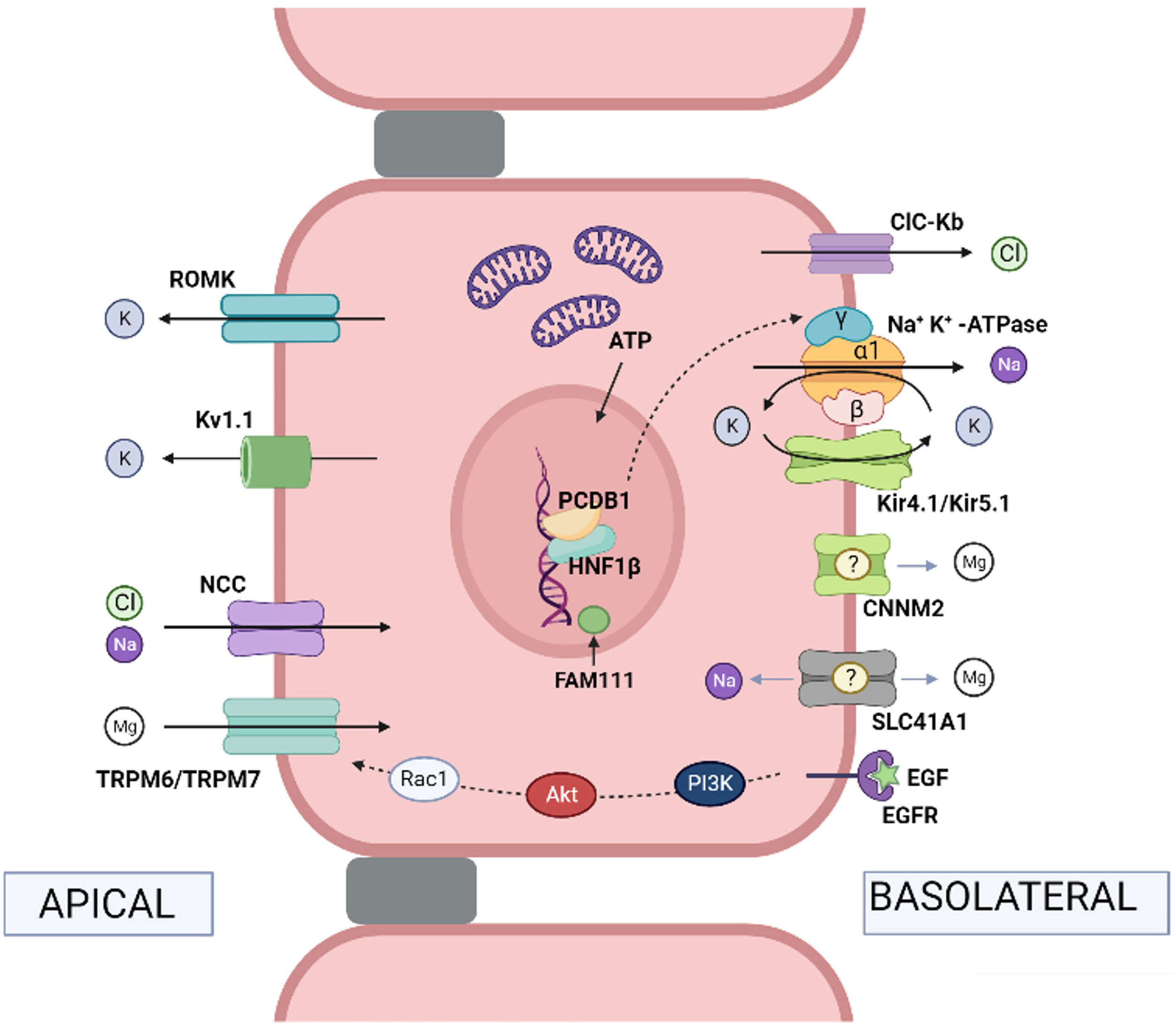
![Physiological mechanisms of transport in the distal convoluted tubule. Na+ and Cl− pass from the tubular lumen into the cell via the thiazide-sensitive NaCl cotransporter (NCC) (mutations in the coding gene cause Gitelman syndrome). Cl− leaves the cell via the chloride channel ClC-Kb (mutations in the gene lead to Bartter syndrome type 3). Na+ leaves the cell via Na+, K+-ATPase (mutations in the gene encoding the ɣ subunit of Na+, K+-ATPase cause autosomal dominant hypomagnesemia with hypocalciuria and those encoding the α subunit cause hypomagnesemia with seizures and type 2 intellectual disability). Hepatocyte nuclear transcription factor 1 (HNF1β) regulates transcription of the gene encoding the ɣ-subunit of the Na+, K+-ATPase (mutations in the coding gene produce HNF1β nephropathy). The coactivator pterin-4-alpha-carbinolamine dehydratase 1 (PCBD1) is a cofactor of HNF1β and increases the transcriptional activity of theγ subunit of Na+, K+-ATPase (mutations in the coding gene produce transient neonatal hyperphenylalaninemia with primapterinuria). FAM111A is a regulator of certain nuclear transcription factors (heterozygous missense mutations in the coding gene cause Kenny-Caffey syndrome type 2 which can present with hypomagnesemia). In the distal tubule, the reabsorption of Mg2+ is an active transport process by transcellular routes, driven by the potential difference between the tubular lumen and the interior of the cell. Mg2+ ions pass from the tubular lumen to the cytosol via the apical TRPM6/TRPM7 channel (mutations in the TRPM6 and TRPM7 coding genes produce hypomagnesemia with autosomal recessive secondary hypocalcemia type 1 and type 2, respectively). Mg2+ ions may exit the cell via CNNM2 (mutations in the coding gene produce hypomagnesemia with seizures and intellectual disability type 1) and the Mg2+ exchanger/Na+ SLC41A1 (mutations in the coding gene produce a nephronophthisis-like phenotype). Epidermal growth factor (EGF) is an activator of the TRPM6 channel (mutations in the coding gene produce autosomal recessive hypomagnesemia; mutations in the EGF receptor [EGFR] produce neonatal inflammatory skin and bowel disease type 2). The apical force required for Mg2+ transport is created by the cooperative action of the basolateral Na+, K+-ATPase, the chloride channel ClC-Kb, the heteromeric channel Kir4.1/Kir5.1 that recycles K+ (mutations in the gene encoding Kir 4.1 produce EAST/SeSAME syndrome), the NCC and K+ extruding channels, ROMK (Kir1.1) (mutations in the coding gene produce Bartter syndrome type II) and Kv1.1 (mutations in the coding gene produce autosomal dominant hypomagnesemia or episodic ataxia type 1). Note: the names of the coding genes are not included here as they are given in the text.](https://static.elsevier.es/multimedia/20132514/0000004400000001/v1_202403090635/S2013251424000452/v1_202403090635/en/main.assets/thumbnail/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w94GCRvdQBB6xyQjMrWMzrts= "Physiological mechanisms of transport in the distal convoluted tubule. Na+ and Cl− pass from the tubular lumen into the cell via the thiazide-sensitive NaCl cotransporter (NCC) (mutations in the coding gene cause Gitelman syndrome). Cl− leaves the cell via the chloride channel ClC-Kb (mutations in the gene lead to Bartter syndrome type 3). Na+ leaves the cell via Na+, K+-ATPase (mutations in the gene encoding the ɣ subunit of Na+, K+-ATPase cause autosomal dominant hypomagnesemia with hypocalciuria and those encoding the α subunit cause hypomagnesemia with seizures and type 2 intellectual disability). Hepatocyte nuclear transcription factor 1 (HNF1β) regulates transcription of the gene encoding the ɣ-subunit of the Na+, K+-ATPase (mutations in the coding gene produce HNF1β nephropathy). The coactivator pterin-4-alpha-carbinolamine dehydratase 1 (PCBD1) is a cofactor of HNF1β and increases the transcriptional activity of theγ subunit of Na+, K+-ATPase (mutations in the coding gene produce transient neonatal hyperphenylalaninemia with primapterinuria). FAM111A is a regulator of certain nuclear transcription factors (heterozygous missense mutations in the coding gene cause Kenny-Caffey syndrome type 2 which can present with hypomagnesemia). In the distal tubule, the reabsorption of Mg2+ is an active transport process by transcellular routes, driven by the potential difference between the tubular lumen and the interior of the cell. Mg2+ ions pass from the tubular lumen to the cytosol via the apical TRPM6/TRPM7 channel (mutations in the TRPM6 and TRPM7 coding genes produce hypomagnesemia with autosomal recessive secondary hypocalcemia type 1 and type 2, respectively). Mg2+ ions may exit the cell via CNNM2 (mutations in the coding gene produce hypomagnesemia with seizures and intellectual disability type 1) and the Mg2+ exchanger/Na+ SLC41A1 (mutations in the coding gene produce a nephronophthisis-like phenotype). Epidermal growth factor (EGF) is an activator of the TRPM6 channel (mutations in the coding gene produce autosomal recessive hypomagnesemia; mutations in the EGF receptor [EGFR] produce neonatal inflammatory skin and bowel disease type 2). The apical force required for Mg2+ transport is created by the cooperative action of the basolateral Na+, K+-ATPase, the chloride channel ClC-Kb, the heteromeric channel Kir4.1/Kir5.1 that recycles K+ (mutations in the gene encoding Kir 4.1 produce EAST/SeSAME syndrome), the NCC and K+ extruding channels, ROMK (Kir1.1) (mutations in the coding gene produce Bartter syndrome type II) and Kv1.1 (mutations in the coding gene produce autosomal dominant hypomagnesemia or episodic ataxia type 1). Note: the names of the coding genes are not included here as they are given in the text.") Fig. 1.
Fig. 1.Physiological mechanisms of transport in the distal convoluted tubule. Na+ and Cl− pass from the tubular lumen into the cell via the thiazide-sensitive NaCl cotransporter (NCC) (mutations in the coding gene cause Gitelman syndrome). Cl− leaves the cell via the chloride channel ClC-Kb (mutations in the gene lead to Bartter syndrome type 3). Na+ leaves the cell via Na+, K+-ATPase (mutations in the gene encoding the ɣ subunit of Na+, K+-ATPase cause autosomal dominant hypomagnesemia with hypocalciuria and those encoding the α subunit cause hypomagnesemia with seizures and type 2 intellectual disability). Hepatocyte nuclear transcription factor 1 (HNF1β) regulates transcription of the gene encoding the ɣ-subunit of the Na+, K+-ATPase (mutations in the coding gene produce HNF1β nephropathy). The coactivator pterin-4-alpha-carbinolamine dehydratase 1 (PCBD1) is a cofactor of HNF1β and increases the transcriptional activity of theγ subunit of Na+, K+-ATPase (mutations in the coding gene produce transient neonatal hyperphenylalaninemia with primapterinuria). FAM111A is a regulator of certain nuclear transcription factors (heterozygous missense mutations in the coding gene cause Kenny-Caffey syndrome type 2 which can present with hypomagnesemia). In the distal tubule, the reabsorption of Mg2+ is an active transport process by transcellular routes, driven by the potential difference between the tubular lumen and the interior of the cell. Mg2+ ions pass from the tubular lumen to the cytosol via the apical TRPM6/TRPM7 channel (mutations in the TRPM6 and TRPM7 coding genes produce hypomagnesemia with autosomal recessive secondary hypocalcemia type 1 and type 2, respectively). Mg2+ ions may exit the cell via CNNM2 (mutations in the coding gene produce hypomagnesemia with seizures and intellectual disability type 1) and the Mg2+ exchanger/Na+ SLC41A1 (mutations in the coding gene produce a nephronophthisis-like phenotype). Epidermal growth factor (EGF) is an activator of the TRPM6 channel (mutations in the coding gene produce autosomal recessive hypomagnesemia; mutations in the EGF receptor [EGFR] produce neonatal inflammatory skin and bowel disease type 2). The apical force required for Mg2+ transport is created by the cooperative action of the basolateral Na+, K+-ATPase, the chloride channel ClC-Kb, the heteromeric channel Kir4.1/Kir5.1 that recycles K+ (mutations in the gene encoding Kir 4.1 produce EAST/SeSAME syndrome), the NCC and K+ extruding channels, ROMK (Kir1.1) (mutations in the coding gene produce Bartter syndrome type II) and Kv1.1 (mutations in the coding gene produce autosomal dominant hypomagnesemia or episodic ataxia type 1). Note: the names of the coding genes are not included here as they are given in the text.
(0.26MB). - b)
In patients with Gitelman syndrome, the observation that the electrolyte abnormalities resembled the effects produced by chronic administration of thiazides,15 as well as the results obtained in clearance studies,16 suggested that the defect must lie in the distal tubular transport of sodium and chloride sensitive to thiazides. Indeed, in 1996, it was established that Gitelman syndrome is caused by a reduction in NaCl transport in the distal convoluted tubule due to mutations in the SLC12A3 gene encoding the thiazide-sensitive NaCl cotransporter (NCC) located in the luminal membrane of the distal convoluted tubule cells17 (Fig. 1). It has been suggested that hypomagnesemia in this disease is secondary to a reduction in the activity of the epithelial Mg channel2+ TRPM6 also located in the apical membrane of the distal convoluted tubule18 (Fig. 1).
- c)
In 2002, two independent groups demonstrated that hypomagnesemia with secondary hypocalcemia is an autosomal recessive inherited entity caused by mutations in the Transient receptor potential melastatin type 6 (TRPM6) gene.19,20 Soon after, new mutations in this gene were described.21,22 The TRPM6 protein is a Ca2+ and Mg2+ permeable channel expressed in the luminal membranes of the intestinal epithelium and the cells of the distal convoluted tubule and the collecting duct (Fig. 1). Inactivating mutations of TRPM6 combine an impaired intestinal absorption of Mg2+ and excessive renal loss of Mg.23
The hypocalcemia that is associated with hypomagnesemia is primarily due to a decrease in parathormone synthesis and release because of decreased calcium-sensitive receptor (CaSR) activity, as well as increased parathormone receptor resistance in bone tissue. Thus, parathormone levels are inappropriately low for the concentration of serum calcium. Treatment requires high doses of oral Mg2+ if tolerated, as passive, unsaturable paracellular transport of Mg2+ at the level of the intestine is normal.
- d)
In 1999, Simon et al. described the existence of a protein, paracellin-1, which is necessary for paracellular tubular reabsorption of Mg2+.24 This protein exists in the tight junction zones of the ascending thick branch cells of the loop of Henle. This work established that mutations in the PCLN-1 gene encoding paracellin-1 were the cause of familial hypomagnesemia with hypercalciuria and nephrocalcinosis.24 When it was found that paracellin-1 is a member of the claudin family, it was renamed claudin-16 (CLDN16 gene). Shortly after, it was observed that there were subjects with the disease who did not carry mutations in the CLDN16 gene. In this regard, it was striking that Spanish patients, except for a very few,25 did not have mutations in the gene.26 In 2006, Konrad et al. resolved the issue by identifying subjects carrying mutations in a new gene, CLDN19, another member of the multigene family of claudins, who had hypomagnesemia, chronic kidney disease and severe ocular abnormalities.27 The gene product, claudin-19, performs its function, like claudin-16, at the tight junctions of the renal tubule and retina. Under physiological conditions, claudin-19 acts as a selective barrier to cations at tight junctions and regulates permeability to monovalent and divalent cations.28 In 2008, Hou et al. demonstrated that claudin-16 interacts with claudin-19,29 such that this association confers tight junctions the ability to contain a selective mechanism in cation reabsorption.30 Our group demonstrated that most Spanish patients carry the same mutation (p.G20D) located in the CLDN19 gene, which facilitates the molecular diagnosis of the disease in our country.31–33
![Physiological mechanisms of transport in the distal convoluted tubule. Na+ and Cl− pass from the tubular lumen into the cell via the thiazide-sensitive NaCl cotransporter (NCC) (mutations in the coding gene cause Gitelman syndrome). Cl− leaves the cell via the chloride channel ClC-Kb (mutations in the gene lead to Bartter syndrome type 3). Na+ leaves the cell via Na+, K+-ATPase (mutations in the gene encoding the ɣ subunit of Na+, K+-ATPase cause autosomal dominant hypomagnesemia with hypocalciuria and those encoding the α subunit cause hypomagnesemia with seizures and type 2 intellectual disability). Hepatocyte nuclear transcription factor 1 (HNF1β) regulates transcription of the gene encoding the ɣ-subunit of the Na+, K+-ATPase (mutations in the coding gene produce HNF1β nephropathy). The coactivator pterin-4-alpha-carbinolamine dehydratase 1 (PCBD1) is a cofactor of HNF1β and increases the transcriptional activity of theγ subunit of Na+, K+-ATPase (mutations in the coding gene produce transient neonatal hyperphenylalaninemia with primapterinuria). FAM111A is a regulator of certain nuclear transcription factors (heterozygous missense mutations in the coding gene cause Kenny-Caffey syndrome type 2 which can present with hypomagnesemia). In the distal tubule, the reabsorption of Mg2+ is an active transport process by transcellular routes, driven by the potential difference between the tubular lumen and the interior of the cell. Mg2+ ions pass from the tubular lumen to the cytosol via the apical TRPM6/TRPM7 channel (mutations in the TRPM6 and TRPM7 coding genes produce hypomagnesemia with autosomal recessive secondary hypocalcemia type 1 and type 2, respectively). Mg2+ ions may exit the cell via CNNM2 (mutations in the coding gene produce hypomagnesemia with seizures and intellectual disability type 1) and the Mg2+ exchanger/Na+ SLC41A1 (mutations in the coding gene produce a nephronophthisis-like phenotype). Epidermal growth factor (EGF) is an activator of the TRPM6 channel (mutations in the coding gene produce autosomal recessive hypomagnesemia; mutations in the EGF receptor [EGFR] produce neonatal inflammatory skin and bowel disease type 2). The apical force required for Mg2+ transport is created by the cooperative action of the basolateral Na+, K+-ATPase, the chloride channel ClC-Kb, the heteromeric channel Kir4.1/Kir5.1 that recycles K+ (mutations in the gene encoding Kir 4.1 produce EAST/SeSAME syndrome), the NCC and K+ extruding channels, ROMK (Kir1.1) (mutations in the coding gene produce Bartter syndrome type II) and Kv1.1 (mutations in the coding gene produce autosomal dominant hypomagnesemia or episodic ataxia type 1). Note: the names of the coding genes are not included here as they are given in the text.](https://static.elsevier.es/multimedia/20132514/0000004400000001/v1_202403090635/S2013251424000452/v1_202403090635/en/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w92lL8PRnKGMnsiVXV6EVJ5QRkjJZIKj2umwUyY+cL6K8cyMCoG0zLrx/ORKa7YTjKSEwICSsAffP5UQmjXgKlOCdkuc5AToLutJp6GBJ0BGKQb2sPgGrow+b5Vv9OosxHF/7EfeEzMcrFFsLv7JsvjOabpsK2Iexc5kztg/hkkq5JIWpHvCb/Ayzo602GXTCb+Snilrz24hCkKvpvlQbSik6DaC1Mkdcp24HLQsP073lQdYZWtarBVYud+jTD8Z/jO/ngc/gQVBZewmZnIirGyJXYKAIpltl+zq3dupX0Lzz "Physiological mechanisms of transport in the distal convoluted tubule. Na+ and Cl− pass from the tubular lumen into the cell via the thiazide-sensitive NaCl cotransporter (NCC) (mutations in the coding gene cause Gitelman syndrome). Cl− leaves the cell via the chloride channel ClC-Kb (mutations in the gene lead to Bartter syndrome type 3). Na+ leaves the cell via Na+, K+-ATPase (mutations in the gene encoding the ɣ subunit of Na+, K+-ATPase cause autosomal dominant hypomagnesemia with hypocalciuria and those encoding the α subunit cause hypomagnesemia with seizures and type 2 intellectual disability). Hepatocyte nuclear transcription factor 1 (HNF1β) regulates transcription of the gene encoding the ɣ-subunit of the Na+, K+-ATPase (mutations in the coding gene produce HNF1β nephropathy). The coactivator pterin-4-alpha-carbinolamine dehydratase 1 (PCBD1) is a cofactor of HNF1β and increases the transcriptional activity of theγ subunit of Na+, K+-ATPase (mutations in the coding gene produce transient neonatal hyperphenylalaninemia with primapterinuria). FAM111A is a regulator of certain nuclear transcription factors (heterozygous missense mutations in the coding gene cause Kenny-Caffey syndrome type 2 which can present with hypomagnesemia). In the distal tubule, the reabsorption of Mg2+ is an active transport process by transcellular routes, driven by the potential difference between the tubular lumen and the interior of the cell. Mg2+ ions pass from the tubular lumen to the cytosol via the apical TRPM6/TRPM7 channel (mutations in the TRPM6 and TRPM7 coding genes produce hypomagnesemia with autosomal recessive secondary hypocalcemia type 1 and type 2, respectively). Mg2+ ions may exit the cell via CNNM2 (mutations in the coding gene produce hypomagnesemia with seizures and intellectual disability type 1) and the Mg2+ exchanger/Na+ SLC41A1 (mutations in the coding gene produce a nephronophthisis-like phenotype). Epidermal growth factor (EGF) is an activator of the TRPM6 channel (mutations in the coding gene produce autosomal recessive hypomagnesemia; mutations in the EGF receptor [EGFR] produce neonatal inflammatory skin and bowel disease type 2). The apical force required for Mg2+ transport is created by the cooperative action of the basolateral Na+, K+-ATPase, the chloride channel ClC-Kb, the heteromeric channel Kir4.1/Kir5.1 that recycles K+ (mutations in the gene encoding Kir 4.1 produce EAST/SeSAME syndrome), the NCC and K+ extruding channels, ROMK (Kir1.1) (mutations in the coding gene produce Bartter syndrome type II) and Kv1.1 (mutations in the coding gene produce autosomal dominant hypomagnesemia or episodic ataxia type 1). Note: the names of the coding genes are not included here as they are given in the text.")
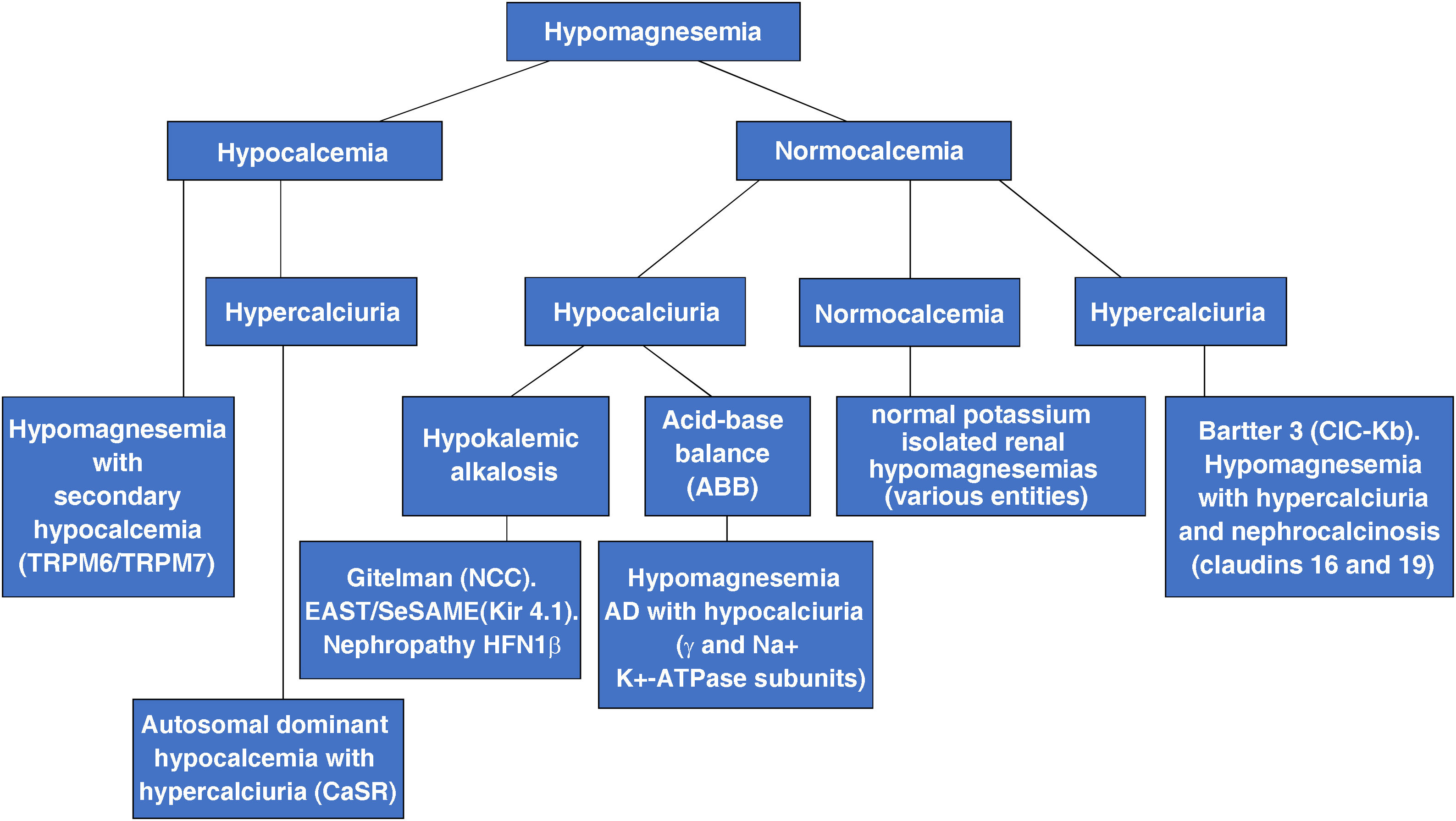
At present, clinicians know of more than 15 genetic causes of renal tubular hypomagnesemia. According to biochemical criteria they can be classified into three subtypes12:
Tubulopathies with hypokalemia and metabolic alkalosis. The anomalous protein can be in the thick ascending branch of the loop of Henle and/or the distal convoluted tubule. This subgroup includes, among others, Bartter syndrome type 3,1,13,14 Gitelman syndrome,2,15–18 seizure syndrome, sensorineural deafness, ataxia, intellectual disability, tubulopathy (EAST/SeSAME)34–36 and HFN1β nephropathy (malformations of the urinary and genital tracts, renal cysts, gout, urinary Mg2+ loss, elevated liver enzymes and juvenile diabetes type MODY).37–39 Renal biochemical data of EAST/SeSAME syndrome and HFN1β nephropathy are similar to those of Gitelman syndrome, including hypocalciuria.
Tubulopathies with hypercalciuria. This subgroup includes entities in which the abnormal proteins are expressed in the thick ascending branch of the loop of Henle. We refer to the two variants of familial hypomagnesemia with hypercalciuria and nephrocalcinosis6–11,24–33 and the heterozygous “gain-of-function” mutations in the CASR gene encoding the calcium-sensitive receptor CaSR that produce autosomal dominant hypocalcemia with hypercalciuria.40,41 In this case, the parathyroid CaSR becomes more sensitive and will detect hypocalcemia as normal calcium and will not stimulate parathyroid hormone secretion. Hypocalcemia is mild to moderate and may be symptomatic in about half of cases (paresthesias, carpopedal spasm and seizures). Activation of CaSR present in the basolateral membrane of the ascending limb of Henle’s loop inhibits the activity of the luminal ROMK channel (Kir 1.1) and produces a pseudo-Bartter’s syndrome. Secondarily, NaCl reabsorption and cation permeability of the claudin-16/claudin-19 channel are reduced with the consequence of salt loss, hypercalciuria, hypermagnesuria and activation of the renin-angiotensin-aldosterone system. Hypokalemic alkalosis may be present.
Tubulopathies without hypercalciuria or hypokalemic metabolic alkalosis. In this subgroup, the abnormal proteins are expressed in the distal convoluted tubule. They are rare isolated hypomagnesemias that, in some cases, present with neurological symptoms.42–46 In addition to hypomagnesemia with secondary hypocalcemia3–5,19–23 this subgroup includes, among others, autosomal dominant hypomagnesemia with hypocalciuria characterized by dysfunction of the ɣ-subunit of the Na+, K+-ATPase caused by mutations in the FXYD2 gene,42,43autosomal recessive hypomagnesemia caused by mutations in the epidermal growth factor (EGF) gene44,45 and hypomagnesemia with seizures and intellectual disability type 1 caused by mutations in the CNNM2 gene encoding cyclin M2.46
Multisystemic tubulopathies. This subgroup includes some diseases caused by mutations in mitochondrial genes in which there may also be urinary loss of Mg2+. This is the case for Kearns-Sayre syndrome (progressive external ophthalmoplegia, retinitis pigmentosa, hearing loss, cerebellar ataxia and heart block with clinical debut before the age of 20 years) and for HUPRA syndrome: hyperuricemia (HU), pulmonary hypertension (P), renal failure (R) and metabolic alkalosis (A) (SARS2 gene).
Fig. 2 shows a diagnostic algorithm for hypomagnesemias constructed based on calcium and calciuria levels.
 are shown in parentheses in most cases.")
Transport modalities and interaction between transporters involved in Mg2+ reabsorption in the distal convoluted tubule (Fig. 1).
Mg2+transporters. The main one is the epithelial Mg2+ channel TRPM6 which, as indicated, is located in the apical membrane of the distal convoluted tubule and brings Mg2+ from the tubular lumen into the cytosol.19–23 Mg2+ is thought to exit the cell through the action of the Mg2+ exchanger/sodium solute carrier family 41, member 1 (SLC41A1) in the basolateral membrane. Mutations in the SLC41A1 gene result in a nephronophthisis-like.47–49 Loss of Na+, K+-ATPase activity hinders the activity of the Mg2+ exchanger -Na+ as Mg2+ extrusion on the basolateral side is dependent on the Na gradient+. Cyclin M2 (CNNM2) is a candidate for basolateral Mg2+ outflow or a regulator of Mg2+ transport but has not yet been confirmed experimentally.46
A regulator of Mg2+channel activity TRPM6. EGF encoded by the EGF gene44,45 binds with high affinity to its receptor EGFR (encoded by the EGFR gene) located on the basolateral membrane of the distal convoluted tubule thereby initiating a signaling cascade for Akt-mediated activation of Rac1, resulting in an increase in TRPM6 channels.
Channels and proteins involved in Mg2+transport by favoring NaCl reabsorption. Mg2+ reabsorption benefits from the generation of a gradient in the apical membrane that enables the NCC cotransporter to transport sodium to the cytosol. Conversely, under pathological conditions, if sodium outflow at the basolateral membrane is reduced, the intracellular Na+ concentration rises and the activity of the NCC transporter is inhibited and, secondarily, that of the Mg2+ channel TRPM6 is reduced. The Na+ pump, K+-ATPase, the Cl channel– ClC-Kb and the heteromeric channel Kir4.1/Kir5.1 (Kir: inwardly rectifying potassium) are all involved in the basolateral membrane. The proper function of the α50 and ɣ42,43 subunits of the Na+, K+-ATPase pump is necessary for Mg2+ reabsorption. Kir4.1/Kir5.1 is a rectifying channel of the intracellular potassium concentration that participates in a K+ recycling mechanism together with the Na+–K+–ATPase (“pump-leak coupling”). An alteration in the function of Kir4.1 favors the uncoupling of the K+ recycling mechanism and reduces the activity of Na+, K+-ATPase, with the consequent inhibition of the activity of the NCC cotransporter.34–36 This is what occurs in EAST/SeSAME syndrome. Mutations have been described in the gene encoding Kir 5.1 that cause the association of distal (hypokalemia, salt loss) and proximal (proximal renal tubular acidosis) tubulopathy with sensorineural deafness; only one of the patients studied had hypomagnesemia.51,52
K+ secretory channels directed toward the tubular lumen are in the apical membrane. Under physiological conditions, potassium that enters the cell via the basolateral Na+, K+-ATPase and escapes the K+ recycling mechanism mentioned above, exits into the tubular lumina through the action of Kv1.1 and ROMK channels (Kir 1.1). The voltage-gated K+ channel Kv1.1 encoded by the KCNA1 gene directly regulates the activity of the Mg2+ channel TRPM6. Thus, when Kv1.1 is non-functional the luminal membrane potential is lost and TRPM653,54 activity is reduced.
A transcription factor, a dimerization cofactor and a transcriptional regulator. Hepatocyte nuclear transcription factor 1 (HNF1β) encoded by the hepatocyte nuclear factor-1β gene (TCF2)37,38 regulates the transcription of the FXYD2 gene encoding the ɣ subunit of the Na+, K+-ATPase which, on the basolateral side, extracts Na+ from the cell in exchange by K+. It is a subunit that adapts the functional properties of this pump to the cellular requirements.42,43
The coactivator pterin-4-alpha-carbinolamine dehydratase 1 (PCBD1) is an HNF1β dimerization cofactor that increases the transcriptional activity of the Na+, K+-ATPase subunitγ. Several mutations in the PCBD1 gene cause reduced activity of the promoter of the FXYD2 gene and lead to renal Mg2+ loss and hypomagnesemia.55 Its association with transient neonatal hyperphenylalaninemia and primapterinuria has been described in a benign neonatal case without long-term sequelae.
FAM111A, a nuclear trypsin-like serine protease, is involved in the regulation of PTH production, calcium homeostasis and bone development and growth. Its nuclear localization suggests that it may be involved in transcriptional regulation. Heterozygous missense mutations in the FAM111A gene that cause Kenny-Caffey syndrome type 2 result in hyperactivation of its intrinsic protein activity that may cause abnormal degradation of DNA-binding proteins.56,57 Kenny-Caffey syndrome is a rare dysmorphological picture characterized by short proportionate stature with heights below 150 cm at adulthood, cortical thickening and medullary stenosis of tubular bones, delayed closure of the anterior fontanel, ocular abnormalities, hypomagnesemia, hypoparathyroidism and hypocalcemia.58 Hypomagnesemia in patients with Kenny-Caffey syndrome type 2 could be attributed to degradation of transcription factors involved in Mg2+ homeostasis.12
Description of a new hypomagnesemic tubulopathyTRPM6 interacts specifically with its closest homologue, the Mg2+-permeable cation channel Transient receptor potential melastatin 7 (TRPM7), resulting in the assembly of functional TRPM6/TRPM7 complexes forming heterotetramers at the luminal cell membrane.59 So far, it had been reported that the TRPM7 channel promotes neuronal death by non-glutamate-dependent calcium overload during ischemic hypoxia injury. Furthermore, a model of cerebral ischemia in experimental animals60 established the relationship between the TRPM7 channel and tissue injury.
As indicated, hypomagnesemia with secondary hypocalcemia of autosomal recessive inheritance is related to mutations in the TRPM6 gene, but, until now, no variants in the TRPM7 gene had been described in patients with hypomagnesemia.
In an international collaborative work, our group has participated in the study of two families affected by hypomagnesemia and hypocalcemia that were not carriers of mutations in TRPM6 in which it was demonstrated for the first time that they had mutations in the TRPM7 gene. The patients suffered seizures and muscle cramps associated with hypomagnesemia (0.25–0.51 mmol/L; normal: 0.70–1 mmol/L) and hypocalcemia (0.94–1.1 mmol/L; normal: 2.13–2.55 mmol/L). In the first family, a splice site variant induced the incorporation of intron 1 sequences into the TRPM7 messenger RNA (mRNA) and generated a premature stop codon. Fibroblasts from one of the patients showed reduced cell growth. In the second family, a heterozygous missense variant in the pore domain led to decreased TRPM7 channel activity.61
A few weeks later, Lei et al. confirmed our findings. These authors studied a patient with recurrent hemiplegic migraine attacks accompanied by “intractable hypomagnesemia” (0.41−0.54 mmol/L). The fractional urinary excretion of Mg2+ was 9.7 % (normal: <4%) and calcium levels were 1.1 mmol/L. The authors located a heterozygous mutation in the TRPM7 gene that produces a variant in the transmembrane region of the TRPM7 protein that is possibly crucial for the normal function of the ion channel.62 Both in this case and in the second family included in our publication these were de novo mutations. Since both cases are heterozygous variants, we believe that this is a new tubulopathy, namely hypomagnesemia with secondary hypocalcemia type 2, possibly of autosomal dominant inheritance.
FinancingThis work was funded by project PI20/00652, integrated in the National R&D&I Plan 2013–2016 and co-funded by the ISCIII-Subdirectorate General for Evaluation and Promotion of Research and the European Regional Development Fund “A way to make Europe”.
Conflict of interestThe authors declare that they have no conflicts of interest.



