
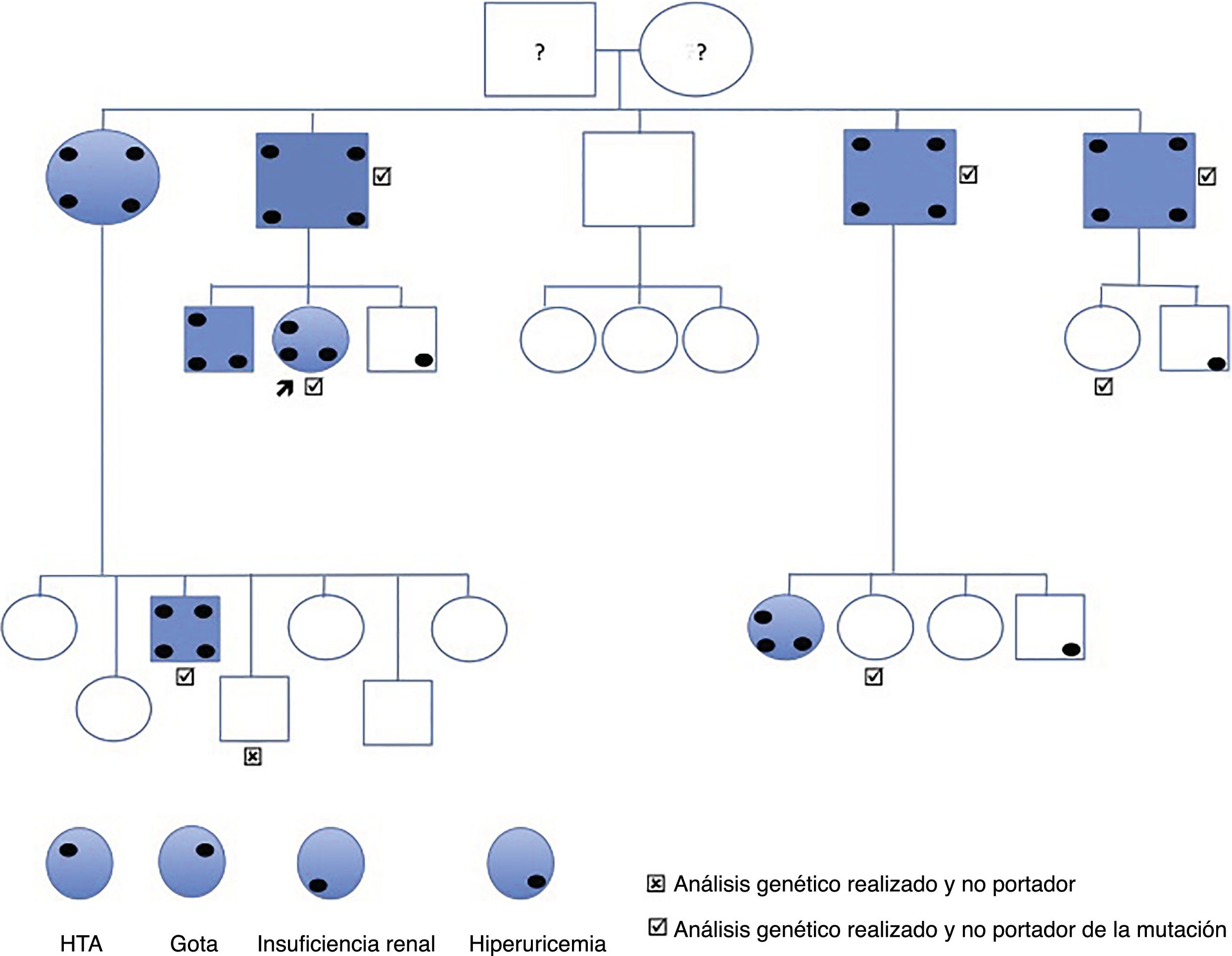
Familial nephropathy associated to hyperuricemia (FJHN) is a group of chronic tubulointerstitial nephritis caused by the alteration of the uromodulin gene (UMOD), and accounts for 1% of advanced chronic kidney disease (ACKD) requiring renal replacement therapy (TRS). We present the case of a family with a mutation of the UMOD gene that has not been previously described.
The index case is a 44-year-old woman referred for high creatinine levels. His background includes smoking, bulbar ulcer H. pylori + and asymptomatic hyperuricemia. She is not on regular medications and does not present any associated extrarenal symptoms. She is diagnosed of grade 1 HTA dipper pattern. Normal kidney ultrasound, without cystic lesions. The lab test reveals hyperuricaemia of 7.6mg/dl, creatinine 1.26mg/dl, MDRD 46.8ml/min/1.73m2, normal urine and 24h CrCl 58ml/min. Her father and 3 out of 4 paternal uncles had kidney failure diagnosed at late ages and at an advanced stage of CKD, one in stage 3b and the other 2 in stage 4 and 5 respectively (Fig. 1).

The rest of first and second degree relatives were evaluated and the findings were the following:
- –
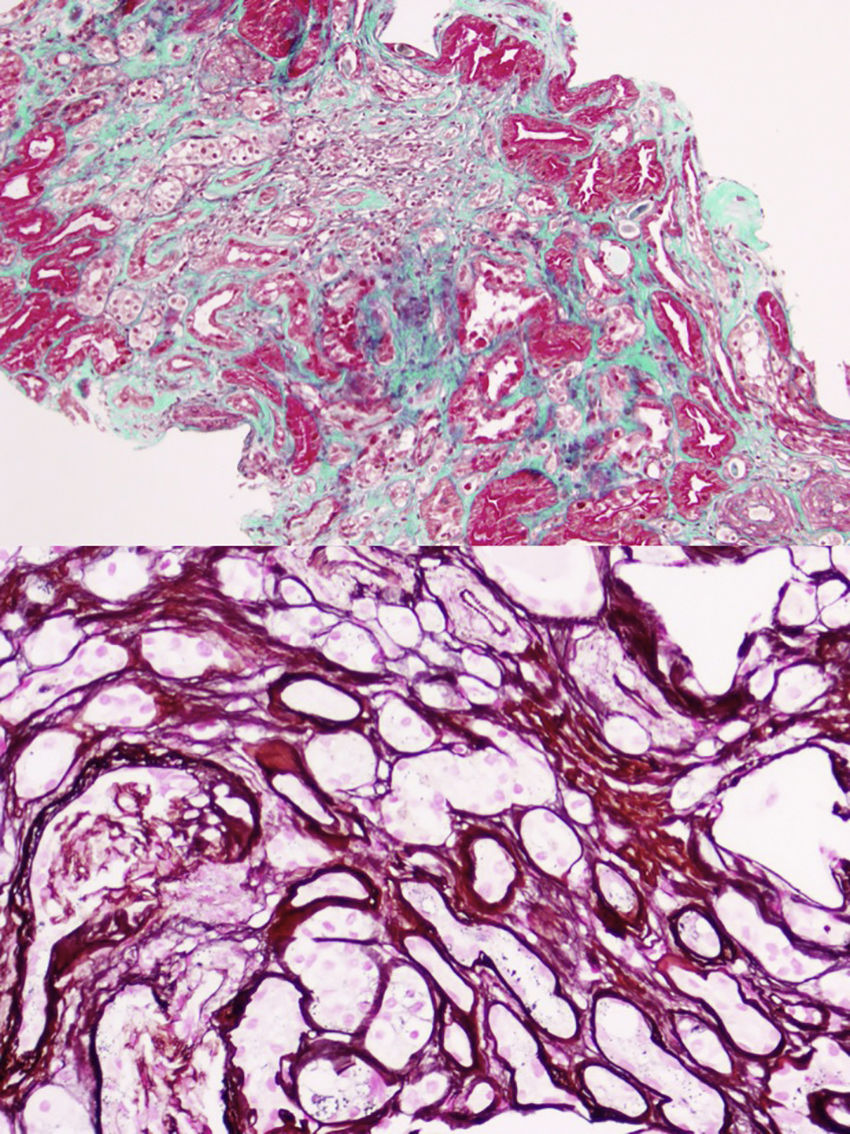
The first generation consists of 5 individuals (father and 4 uncles of the case). All those who have kidney failure also have hyperuricemia. Currently, 2 are kidney transplant recipients and 2 are in advance CKD. In all of them except one, renal ultrasonography is normal without evidence of cystic lesions. A renal biopsy was performed on the only paternal uncle with only one corticomedullary cyst and with advanced CKD MDRD-4 20ml/min/1.73m2, histology showed chronic tubulointerstitial nephropathy (Fig. 2).
- –
The second generation includes the patient case, 2 brothers and 16 cousins. Kidney failure is detected in 4 cases including the patient, 3 in stage 3a and one in stage 2, all of them with hyperuricemia, and 3 cases had hyperuricemia without renal failure. Only one cousin, without renal failure or hyperuricemia, presents corticomedullary cysts.
- –
The third generation consists of 34 people born between 1981 and 2013, all of them studied with normal blood and urine tests

In general we have the following findings:
- –
Renal insufficiency in 4/5 ascendants, 1 out of the 2 brothers and 2 cousins.
- –
Hyperuricemia with hypouricosuria in all affected with renal failure. In addition, urinary osmolarity <500mOsm/kg and hypomagnesuria with normomagnesemia in some of the affected.
- –
Corticomedullary cysts were found only in 2 of the family members studied, and only one had obvious clinical-analytic findings. None presented symptoms or ultrasound findings of renal lithiasis.
- –
Gout only in the 4 patients affected from the first generation.
- –
In all cases HTN coincides with renal failure.
After clinical evaluation it was performed sequencing of exons 2–12 (coding) as well as the flanking intronic bases in one of the patients affected. To this end, fragments were amplified by PCR followed by automatic sequencing using the Sanger method. A new mutation was identified in exon 4: c.517C>G, p.P173A, which was determined in all the family members studied genetically (Fig. 1), all affected were heterozygous carriers. The family member without the mutation did not have any clinical expression or laboratory abnormalities related to the disease. Of the other 6 that do have the mutation, 4 have developed the disease (hyperuricemia and renal failure), while the other 2 did show evidence of the disease.
With all these findings, we make the diagnosis of familial chronic interstitial nephropathy with hyperuricemia caused by the UMOD gene, a variant of type 2 medullary cystic disease.
Mutations in the UMOD gene are the cause of different hereditary hyperuricemic and hypouricosuric nephropathies that can cause, in addition to gout and lithiasis, arterial hypertension and chronic tubulointerstitial nephritis leading to advanced CKD.1,2 The FJHN of dominant autonomic inheritance usually manifests itself in adulthood due to hyperuricemia, and there is a great heterogeneity of the intra- and interfamilial genotype–phenotype of this entity, a situation that also occurs in the family that has been exposed.3,4
Dozens of mutations have been described in the UMOD gene.5,6 In our case, a previously undescribed mutation has been discovered that consists in a modification of the amino acid cytosine by guanine (c.517C>G p.pro173Ala) in the studied fragment that corresponds to exon 3 of the UMOD gene, and that is why we bring it to the attention of the scientific community. Most of the mutations have been located in exon 4,7 but never the current mutation that conditions the amino acid change described above. This newly described mutation would not imply pathogenic character had it not been found in several affected members of the family (Fig. 2). This finding has allowed the preclinical diagnosis of 2 relatives of 39 and 40 years, and in one individual the disease was ruled out. Since all those affected have the mutation, but there are carriers without clinical expression of the disease, we understand that the mutation is related to a variable penetrance, which could depend on the age. Although the progression of renal failure in this entity is slow, its silent nature justifies the monitoring of asymptomatic young relatives, since, although there is no specific therapy, early diagnosis would allow early treatment of indolent hyperuricemia and global recommendations for nephroprotection in an attempt to slow down the onset or progression of the nephropathy.8–10
We conclude that the early detection of FJHN can lead to early treatment that delays the onset or progression of renal failure. The family study and genetic analysis in this disease are important for its definitive diagnosis since the clinic and the histopathology are not specific.
Please cite this article as: Martín-Gómez MA, Eliecer C, Caba Molina M, González Oller C, García del Moral R. Nefropatía familiar hiperuricemiante: nueva mutación familiar del gen de la uromodulina. Nefrologia. 2019;39:309–311.



