
La poliquistosis renal autosómica dominante (PQRAD) es una enfermedad hereditaria, para cuyo diagnóstico no es necesario realizar biopsia renal. Se caracteriza por la presencia de múltiples quistes renales bilaterales que aumentan el tamaño renal y con posibles manifestaciones extrarrenales. En algunos casos, los pacientes afectos de PQRAD presentan proteinuria, pero esta habitualmente es leve y no suele exceder de 1 g/24 horas. En los casos en los que la proteinuria alcanza el rango nefrótico, debe plantearse la posibilidad de otra nefropatía coexistente y, en consecuencia, se ha de realizar una biopsia renal para filiar el diagnóstico, siendo la lesión anatomopatológica más documentada la glomeruloesclerosis focal y segmentaria. A continuación, describimos un caso de una paciente con PQRAD diagnosticada previamente, que presenta durante su primer embarazo un síndrome nefrótico por una glomerulonefritis por cambios mínimos asociada a anticuerpos antinucleares.
CASO CLÍNICO
Mujer caucásica de 29 años, con antecedente de asma en la infancia en tratamiento con salbutamol a demanda y poliquistosis renal autosómica dominante (PQRAD), con función renal conservada, diagnosticada dos años antes por estudio ecográfico solicitado por antecedentes familiares de PQRAD (madre en hemodiálisis y hermana también afecta). Durante su primer embarazo, a las 23 semanas, presenta síndrome nefrótico clínico con edemas en ambas extremidades inferiores, sin edema facial ni hipertensión. Niega síndrome miccional, fiebre, disminución de la diuresis, rash cutáneo, úlceras ni artralgias. La exploración física era anodina, a excepción de los edemas. En la analítica destaca proteinuria de 6,14 g/24 horas y microhematuria, función renal conservada, albúmina sérica de 29,2 g/l, con ionograma normal y hemoglobina de 12,1 g/dl. Se inicia tratamiento con tiacidas que la paciente abandona posteriormente y se realiza ecografía abdominal que confirma poliquistosis hepatorrenal, con riñón derecho de 14 cm y riñón izquierdo de 15 cm a expensas de múltiples quistes corticales bilaterales, con correcta diferenciación córticomedular y córtex renal de grosor normal, sin dilatación de la vía excretora. El estudio inmunológico resulta normal para las fracciones del complemento: C3 147 mg/dl (normalidad 90-180 mg/dl) y C4 24 mg/dl (normalidad 10-40 mg/dl) y con anticuerpos anti-nucleares (ANA) 1/320 y anti-DNA negativo. Dada la aparición de ANA positivo, se repitió el estudio inmunológico, que mostró ANA 1/640 con patrón moteado grueso y con anticuerpos extraíbles del núcleo (ENA) negativos.
En la semana 27 de embarazo requirió ingreso por amenaza de parto prematuro con polihidramnios y malformación fetal (oclusión intestinal), y recibió dos dosis de corticoides intramusculares para maduración pulmonar fetal. En la semana 37 se realizó cesárea electiva.
La paciente persiste posparto con fóvea en ambas extremidades inferiores, normotensa, función renal conservada y proteinuria de 6 g/24h y albúmina sérica de 17 g/l, por lo cual se decide la realización de una biopsia renal percutánea ecoguiada sin incidencias, extrayéndose un cilindro de corteza renal que contenía 26 glomérulos que muestran un leve incremento de la matriz mesangial sin aumento de la celularidad ni alteraciones en las asas capilares, y no se observan glomérulos con lesiones focales o segmentarias. En las arteriolas observamos moderada hialinosis, mientras que las arterias interlobulares no presentan ninguna alteración. En el compartimento túbulo-intersticial se aprecia un mínimo infiltrado inflamatorio con eosinófilos ocasionales, siendo la fibrosis/atrofia tubular < 5 % de la corteza. Tinción de rojo Congo negativa. Inmunofluorescencia con positividad leve e inespecífica en arteriolas con hialinosis para IgM y C3, siendo el resto negativo.
Diagnosticándose de glomerulonefritis por cambios mínimos con moderada hialinosis arterial, se inició tratamiento corticoideo con prednisona en dosis de 1 mg/kg. A las tres semanas de tratamiento destaca solo una excreción urinaria de albúmina de 131 mg/g de creatinina y una albúmina sérica de 39,1 g/l (normal), y desaparición de los edemas en las extremidades inferiores.
DISCUSIÓN
La PQRAD es una enfermedad hereditaria caracterizada por la presencia de múltiples quistes renales bilaterales que aumentan el tamaño renal y con posibles manifestaciones extrarrenales, como quistes hepáticos, aneurismas intracraneales, insuficiencia mitral y aneurismas de aorta abdominal1. La frecuencia de proteinuria en pacientes con PQRAD se ha descrito entre un 14 y 34 % en aquellos con función renal conservada y hasta en un 80 % de los adultos con enfermedad renal crónica, en la revisión realizada en 1994 por Chapman et al.2. La proteinuria leve (< 1 g/24 horas) es un hallazgo frecuente en las personas con PQRAD. Sin embargo, en el caso de proteinuria severa, debemos sospechar una complicación en forma de enfermedad glomerular, por lo cual, cuando se nos presenta proteinuria de rango nefrótico en un paciente con PQRAD, se ha de plantear la necesidad de realizar una biopsia renal para llegar al diagnóstico de certeza.
La aparición de una enfermedad glomerular coincidente se ha descrito que acelera el curso de la PQRAD a enfermedad renal crónica terminal y que suele requerir medidas específicas para el control de la proteinuria.
Dado que actualmente no disponemos de un tratamiento específico para enlentecer la progresión de la PQRAD, y en vistas del empeoramiento que puede provocar la glomerulopatía añadida, es especialmente importante un diagnóstico y tratamiento adecuados de la enfermedad glomerular sobreañadida.
La biopsia renal es una fuente de información muy importante en cuanto al diagnóstico, pronóstico y tratamiento de los pacientes con enfermedad renal. En el caso de la PQRAD con síndrome nefrótico añadido, es más controvertido, dados los riesgos que entraña la biopsia renal por la propia presencia de quistes renales, pero hay que tener en cuenta la importancia de la información que aporta en cuanto a tratamiento y pronóstico del paciente.
Actualmente, con la biopsia guiada por ecografía, que permite una mejor localización del punto de punción más adecuado, el éxito en la obtención de material es muy elevado y las complicaciones son raras. Siempre hay que tener en cuenta que en algunas situaciones especiales puede comportar un riesgo añadido, como por ejemplo en caso de pacientes monorrenos, con alteraciones de la coagulación o en pacientes poco colaboradores. Por lo tanto, en algunos de estos casos habrá que plantear otras opciones, como la biopsia renal abierta con anestesia general.
Dado que los pacientes con quistes renales suponen un riesgo sobreañadido, habrá que plantear métodos menos agresivos y con menor riesgo de sangrado, como la biopsia transyugular, aunque con menos probabilidad de diagnóstico de certeza por la menor eficacia diagnóstica de la técnica.
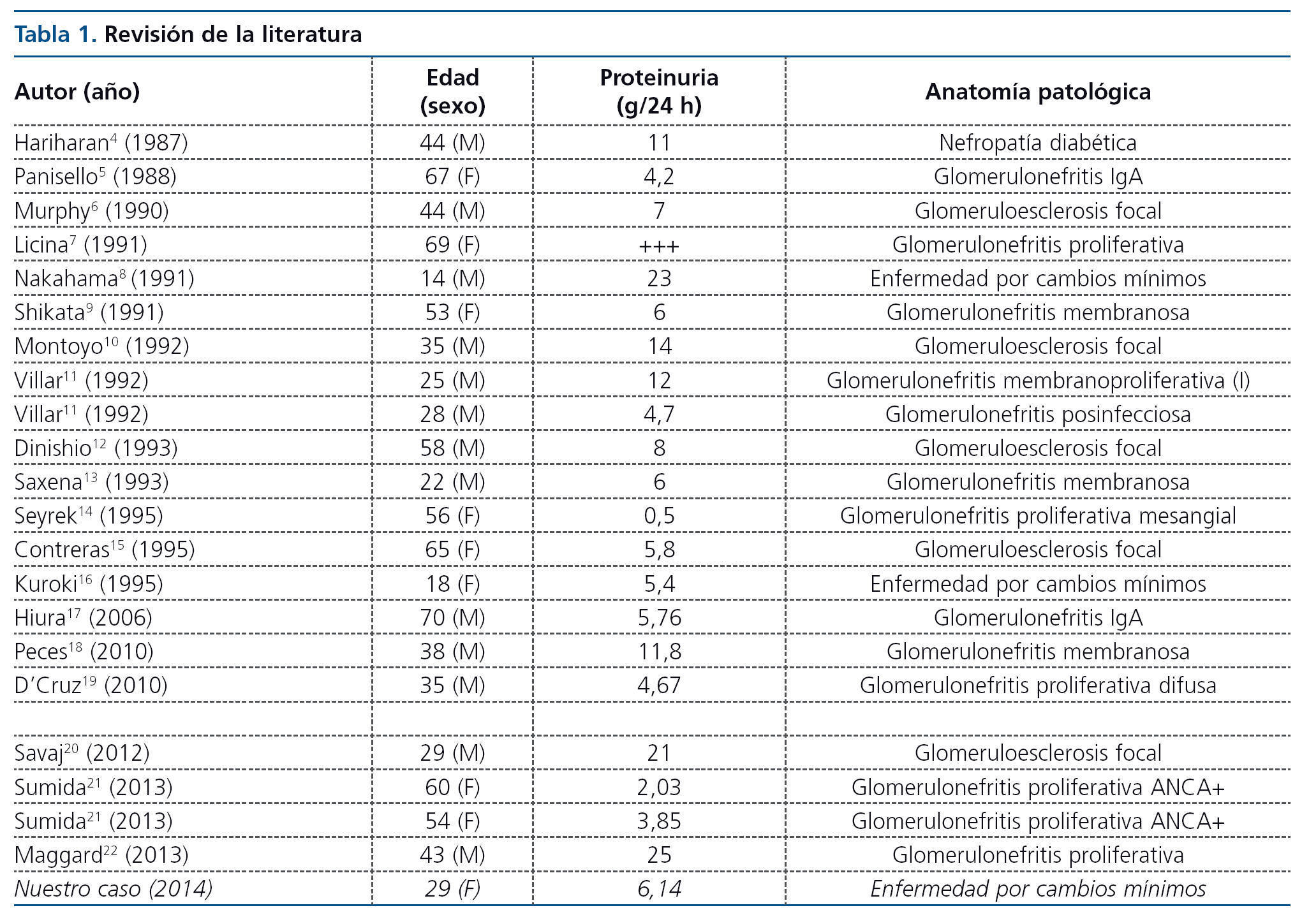
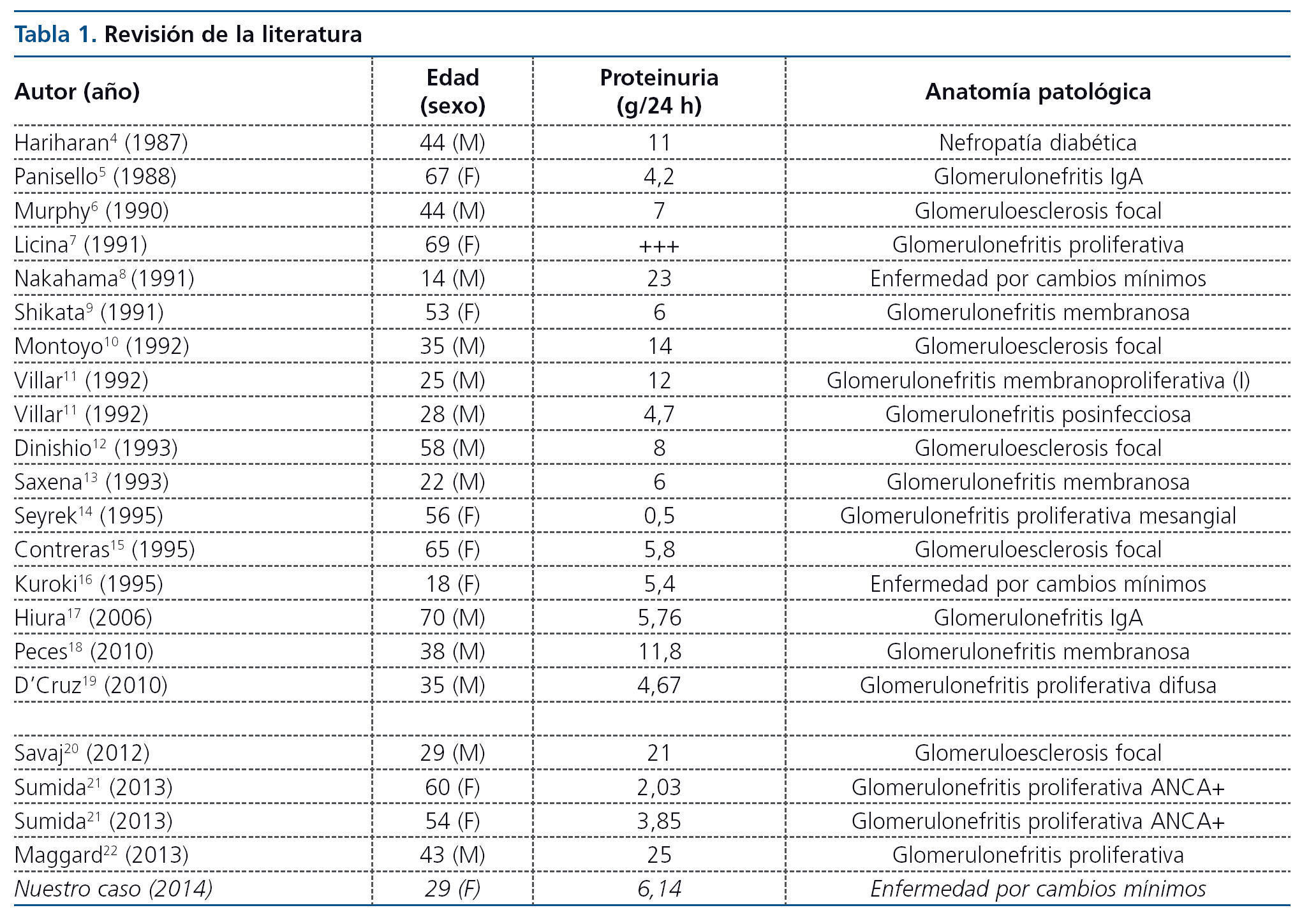
Revisando la literatura publicada, se encuentra únicamente el artículo de Chapman et al.2, donde se realiza una revisión sobre proteinuria asociada a PQRAD, y en él no se hace referencia a las biopsias renales, aunque describe que 48 de los 270 pacientes PQRAD desarrollaron proteinuria > 300 mg/día (un 18 %) y que estos presentaban más prevalencia de hipertensión, riñones de mayor tamaño y progresión más rápida de la enfermedad. Por otro lado, Dalgaard et al.3 realizaron un seguimiento de dos años de 122 pacientes con PQRAD donde se documentaron tres casos de pacientes con proteinuria de > 5 g/24 horas, pero tampoco disponemos de biopsia renal en dichos casos. Lo que sí destaca es que la probabilidad de presentar proteinuria de rango nefrótico a medio plazo en personas con PQRAD es baja (3 casos de 122, que representan un 2,45 % de los casos). Dado que en la literatura únicamente se reflejan case reports, hemos decidido realizar una revisión de los artículos publicados en PubMed de los últimos 25 años, encontrando 22 casos de pacientes con PQRAD y síndrome nefrótico con biopsia renal realizada4-22.
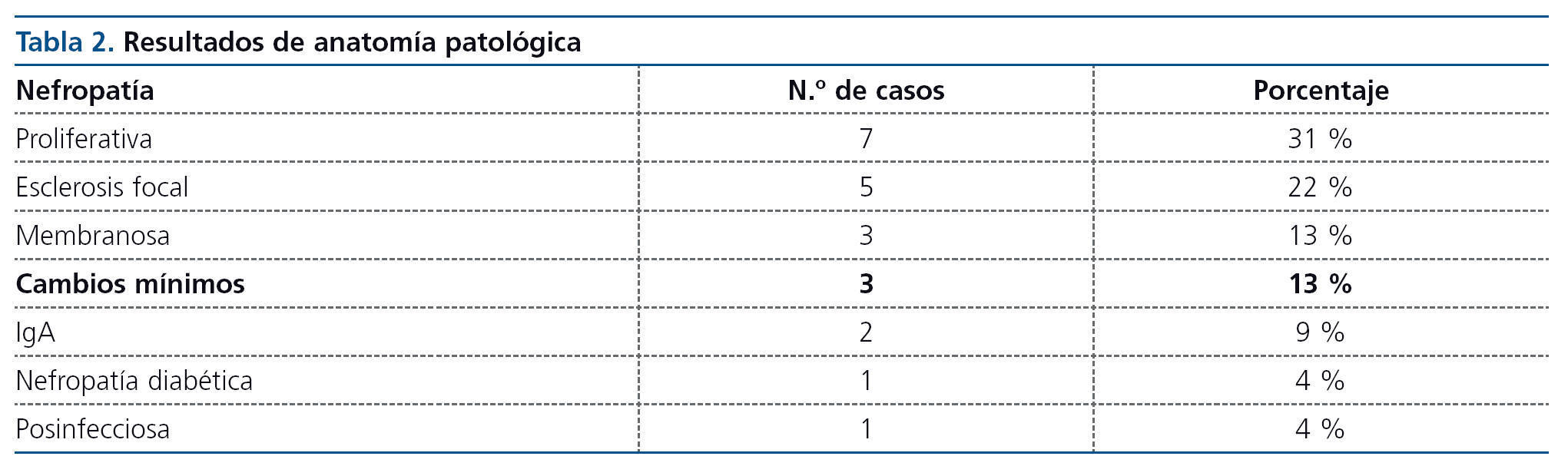
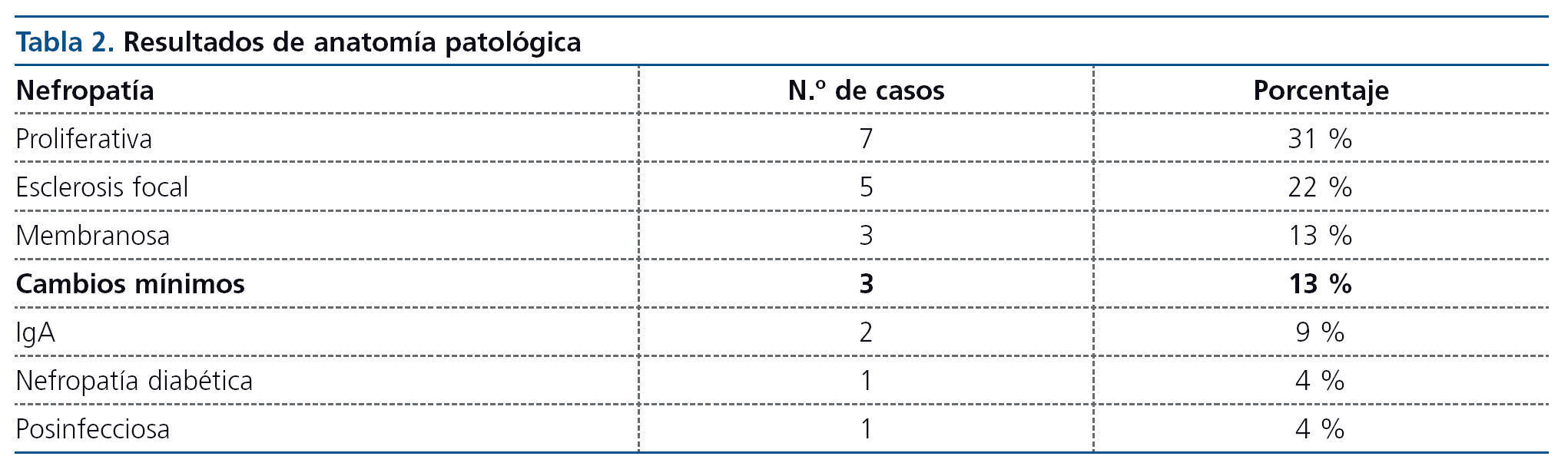
Hay una gran variedad de lesiones histopatológicas en los casos publicados, pero la más frecuente es la glomeruloesclerosis focal y segmentaria, seguida de glomerulonefritis proliferativa (tablas 1 y 2).
En nuestro caso, la paciente presenta glomerulonefritis por cambios mínimos con buena respuesta al tratamiento con corticoides. No se conoce un claro origen de la afectación renal ni si esta asociación es puramente casual o si tiene un mecanismo patogénico concreto.
Nuestra paciente muestra además un ANA positivo con título que oscila entre 1/320 y 1/640 con patrón moteado grueso. Si bien esto nos podría hacer pensar en una enfermedad autoinmune, el hecho de que no presente clínica compatible y que el tipo de patrón sea bastante inespecífico, además de presentar ENA negativos, junto al patrón histológico de la biopsia renal, hacen poco probable la presencia de una enfermedad autoinmune. Aun así, es recomendable el seguimiento clínico y serológico, ya que se han descrito casos en que la presencia de ANA precedía a las manifestaciones clínicas y al resto de manifestaciones analíticas de enfermedad autoinmunitaria, sobre todo en el lupus eritematoso sistémico23.
CONCLUSIONES
Los clínicos son reticentes a realizar una biopsia renal en un paciente con PQRAD a pesar de presentar síndrome nefrótico, dados los riesgos que puede comportar dicha técnica. Aun así, nuestro caso, como los anteriores en la literatura, remarca la necesidad de llevar a cabo la biopsia renal en pacientes con PQRAD que presenten síndrome nefrótico, ya que es la única manera de excluir otras causas o diagnosticarlas y, por consiguiente, tratar el cuadro sobreañadido. En este último aspecto, es importante obtener un diagnóstico concreto basado en la anatomía patológica, ya que, como es sabido, el tratamiento difiere entre las distintas nefropatías posibles y los diferentes estadios de la enfermedad sobreañadida.
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.
doi:10.3265/NefroPlus.pre2015.Feb.12792
Correspondencia:
Mónica Pérez Mir
Servicio de Nefrología. Hospital Universitari Germans Trias i Pujol.
Carretera de Canyet, s/n. 08916 Badalona, Barcelona.
mperezmir@yahoo.com


