
La poliquistosis renal autosómica dominante es la enfermedad renal hereditaria más frecuente aunque los datos disponibles generalmente son tras el inicio del tratamiento renal sustitutivo.
ObjetivoConocer la situación global de la poliquistosis renal autosómica dominante en el ámbito sanitario de Granada.
Material y métodosDesde enero 2007 hasta diciembre 2016 hemos recogido información clínica, familiar y demográfica de todos los pacientes con poliquistosis renal autosómica dominante, estuvieran o no en tratamiento renal sustitutivo, atendidos en el área de Granada. Se han utilizado los programas informáticos SPSS 15.0 y GenoPro.
ResultadosMil ciento siete pacientes diagnosticados, el 50,6% son varones. Se han estudiado 4-6 generaciones/familia. El 99,1% de raza caucásica. Hay áreas geográficas con mayor concentración. No hay antecedentes familiares en el 2,43%. La edad media de diagnóstico es de 34±17,8 años y en el 57,7% de los casos, el diagnóstico se produce después de tener descendencia. El principal motivo de diagnóstico son los antecedentes familiares (46,4%). La edad media de entrada en técnica es de 54,2±11,05 años. El 96,3% de los fallecidos tenían algún grado de insuficiencia renal en el momento del exitus. La edad media del exitus es de 60,9±14,10 años, siendo desconocida la principal causa de muerte (33,5%) seguida de la cardiovascular (27,8%).
ConclusionesCasos y familias se concentran en algunas áreas geográficas, un número importante de individuos están sin diagnosticar, fallecen antes por causa cardiovascular y se diagnostican tarde respecto al momento reproductivo. Dado que no hay tratamiento curativo, la estrategia de prevención primaria mediante el diagnóstico genético preimplantacional adquiere protagonismo.
Although autosomal dominant polycystic kidney disease is the most common hereditary kidney disease, available data tend to be limited to after initiation of renal replacement therapy.
ObjectiveTo ascertain an overview of autosomal dominant polycystic kidney disease within the health area of Granada in southern Spain.
Material and methodsFrom January 2007 to December 2016, we collected clinical, family and demographic information about all patients with autosomal dominant polycystic kidney disease, irrespective of whether or not they were treated with RRT, in the Granada health area. The computer software SPSS 15.0 and GenoPro were used.
Results50.6% of the 1,107 diagnosed patients were men. 99.1% were Caucasian and 4–6 generations/family were studied. The geographical distribution was heterogeneous. There was no family history in 2.43%. The mean age of diagnosis was 34.0±17.80 years and the diagnosis was made after having offspring in 57.7% of cases. The main reason for diagnosis was family history (46.4%). The mean age of initiation of renal replacement therapy was 54.2±11.05 years. 96.3% of the deceased had some degree of renal failure at the time of death. The mean age of death was 60.9±14.10 years, the main cause of death being unknown in 33.5% of cases, followed by cardiovascular (27.8%).
ConclusionsCases and families were concentrated in certain geographical areas and a significant number of individuals were undiagnosed prior to cardiovascular death or diagnosed late after reproduction. Given that there is currently no curative treatment, the primary prevention strategy of preimplantation genetic diagnosis should play a leading role.
La poliquistosis renal autosómica dominante (PQRAD) es la enfermedad renal hereditaria más frecuente con una prevalencia estimada de 1/800-1.0001; en España esta frecuencia no está bien determinada y desconocemos también si presenta variaciones en el tiempo.
Los pacientes con PQRAD constituyen el 6-10% de la población en diálisis o trasplante renal, siendo una enfermedad con un gran impacto social2,3. En nuestro país es la sexta causa que conduce a tratamiento renal sustitutivo (TRS) según los datos del Registro Español4. Esta misma posición ocupa en Andalucía, de acuerdo con la información recogida en el del Sistema de Información de la Coordinación Autonómica de Trasplantes (SICATA)5. A día de hoy solo se dispone de datos de la enfermedad una vez que los pacientes precisan de TRS en cualquiera de sus modalidades (trasplante, hemodiálisis [HD] y diálisis peritoneal [DP]) pero desconocemos la magnitud global del problema, tanto a nivel provincial como regional y nacional, puesto que no existen registros de aquellos pacientes que no están en TRS. En 2016, la Sociedad Española de Nefrología puso en marcha el registro nacional de PQRAD.
Esta enfermedad se inicia desde el momento de la concepción y va evolucionando a lo largo de la vida hasta que se diagnostica, generalmente por pruebas de imagen, ante una sintomatología sugerente. Estos pacientes presentan síntomas antes de que el deterioro de función renal esté establecido (dolores abdominales, complicaciones hemorrágicas y/o infecciosas de los quistes e hipertensión arterial [HTA]) lo que genera atención médica, tanto en atención primaria como en especializada, que no está cuantificada.
El objetivo principal de nuestro trabajo es analizar la situación epidemiológica de la PQRAD en nuestra región con el fin de identificar y diseñar estrategias sanitarias de prevención innovadoras.
Material y métodosDesde enero 2007 hasta diciembre 2016 hemos recogido información clínica, familiar y demográfica de todos los pacientes diagnosticados de PQRAD, estuvieran o no en TRS, que han sido atendidos en alguna de las instituciones sanitarias de la provincia de Granada y área de referencia. Para ello hemos recabado información de los Servicios de Documentación de los Hospitales San Cecilio y Virgen de las Nieves de Granada, los archivos propios de los servicios de nefrología, los centros de HD concertados con el Servicio Andaluz de Salud, así como de los servicios de Urología, Pediatría, Digestivo, Neurología, Cardiología, Medicina Interna y Genética de ambos hospitales; también de consultas de atención primaria y de médicos y hospitales privados. Para la recogida de datos, estudiar la enfermedad en todos sus aspectos y potenciar las medidas preventivas se creó el Grupo de Estudio de la Enfermedad Poliquística Autosómica Dominante (GEEPAD), formado por un grupo multidisciplinar de profesionales de la salud que de manera directa o indirecta tenían contacto con pacientes con esta enfermedad.
A todos los pacientes vivos se les ha solicitado consentimiento informado. Se les han realizado árboles genealógicos y los datos de los mismos se han almacenado en el programa informático GenoPro.
La información demográfica, familiar, genética y clínica, en total 238 variables, se ha ido introduciendo en una base de datos creada con el programa estadístico SPSS. Para este artículo hemos utilizado las siguientes variables: localidad de nacimiento, domicilio actual, sexo, raza, edad de diagnóstico, si fue antes o después de tener el primer hijo, número de descendientes, cuantía de hijos afectados y de los no estudiados, motivo del diagnóstico, vía de transmisión de la enfermedad, estudio genético, necesidad de TRS, tipo de TRS, afectación de otros órganos y localización, HTA, infecciones y situación del paciente en el momento actual, causa de exitus y si presentaba algún deterioro de la función renal en ese momento.
Para el diagnóstico se han utilizado los criterios unificados de Ravine-Pei6–8.
ResultadosHemos identificado 1.107 pacientes con diagnóstico de PQRAD. Los resultados epidemiológicos más relevantes se resaltan en la tabla 1.
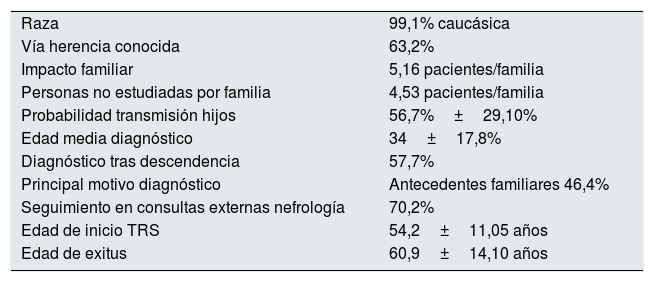
Resultados epidemiológicos más destacados
| Raza | 99,1% caucásica |
| Vía herencia conocida | 63,2% |
| Impacto familiar | 5,16 pacientes/familia |
| Personas no estudiadas por familia | 4,53 pacientes/familia |
| Probabilidad transmisión hijos | 56,7%±29,10% |
| Edad media diagnóstico | 34±17,8% |
| Diagnóstico tras descendencia | 57,7% |
| Principal motivo diagnóstico | Antecedentes familiares 46,4% |
| Seguimiento en consultas externas nefrología | 70,2% |
| Edad de inicio TRS | 54,2±11,05 años |
| Edad de exitus | 60,9±14,10 años |
La distribución por sexos es homogénea (50,6% varones). La gran mayoría de raza caucásica (99,1%); raza negra (0,5%) y de etnia gitana (0,4%).
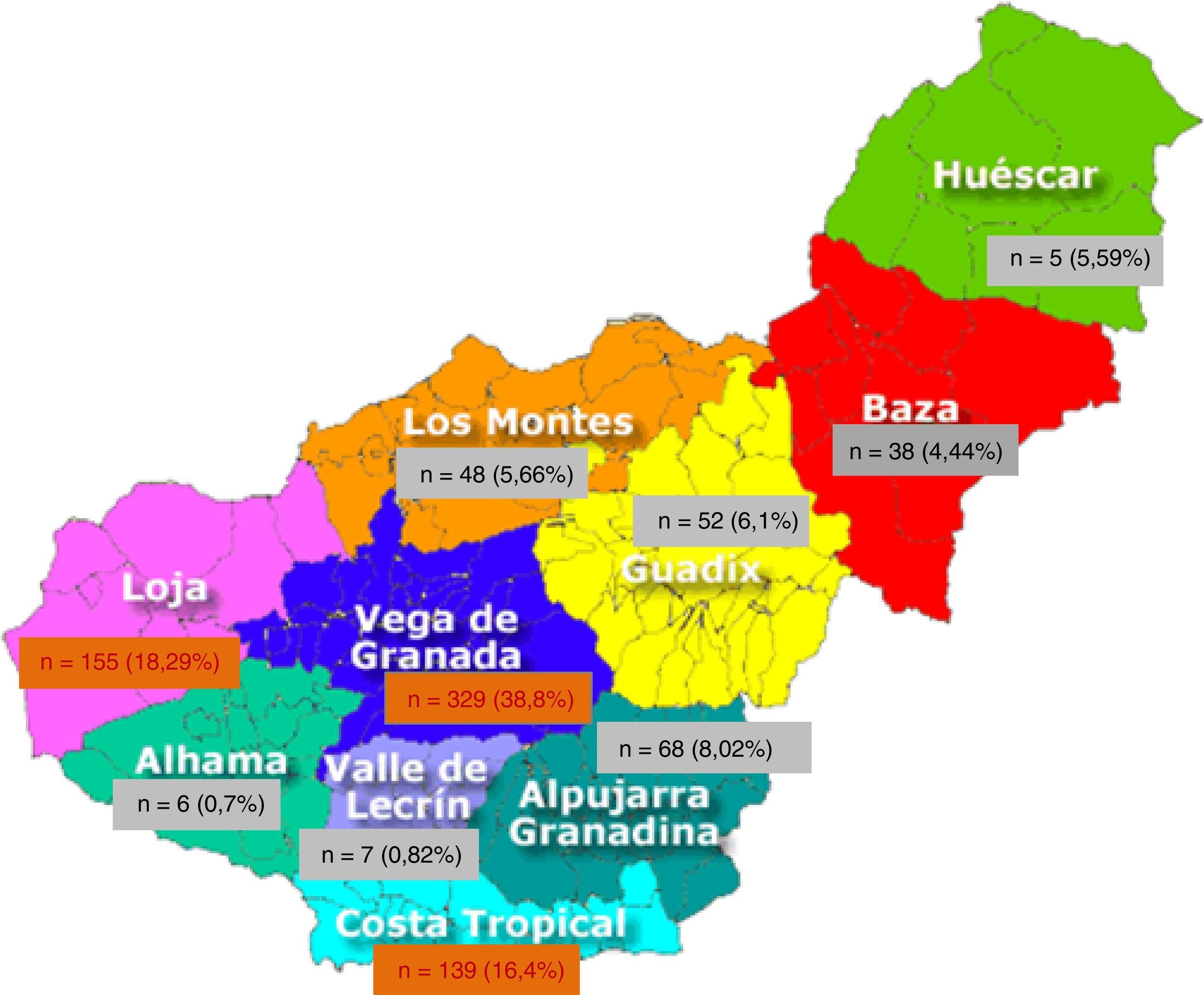
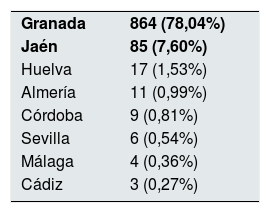
El 99,1% de los pacientes han nacido en Europa y en España el 97,9%, quedando un 2,1% originarios de otros países; 999 (90,24%) han nacido en Andalucía. Por provincias objetivamos que el 78,04% (864 pacientes) son de origen granadino, seguidos de los jiennenses con un 7,6% (tabla 2). De las 10 comarcas de Granada, las más representativas son Vega Granada, Loja y Costa Tropical donde nacieron el 56,2% del total y el 73,5% de la provincia (fig. 1). Granada capital, Íllora y Motril son las 3 ciudades con más número de afectados: 13,7; 5,23 y 4,6%, respectivamente.
Pacientes con PQRAD (n=1107) atendidos en la provincia de Granada, ordenados por provincia andaluza de nacimiento
| Granada | 864 (78,04%) |
| Jaén | 85 (7,60%) |
| Huelva | 17 (1,53%) |
| Almería | 11 (0,99%) |
| Córdoba | 9 (0,81%) |
| Sevilla | 6 (0,54%) |
| Málaga | 4 (0,36%) |
| Cádiz | 3 (0,27%) |
En negrita, la procedencia más frecuente de los pacientes en nuestra área.
.")
En el 63,2% de los casos es clara la vía de herencia: el 33,4% materna, el 29,8% paterna. No identificamos antecedentes familiares en el 2,43%. En el 34,37% no identificamos de forma clara la vía de trasmisión: a) dudosa la materna en el 2,7%, b) dudosa la paterna en el 4,69% y c) desconocida, sin que se pueda descartar un antecedente familiar, en el 26,98%.
Hemos agrupado 986 individuos (89,06%) en 251 familias mediante árboles genealógicos, recogiendo información de 4-6 generaciones/familia. Si seleccionamos aquellos que tienen más de 25 años, el 77,3% tienen descendencia, con una media de hijos de 1,9±1,60. El impacto familiar de la enfermedad, medido como la media de personas con la enfermedad dentro de una misma familia, es de 5,16 pacientes/familia (rango 1-41). La media de personas no estudiadas dentro de una misma familia es de 4,53 (rango 0-38). En nuestra serie, la probabilidad de transmisión a los hijos ha sido del 56,7% (rango 0-100%), sin diferencias según el sexo.
La edad de diagnóstico de la enfermedad es de 34±17,8 años (n=641). En el 57,7% de los casos el diagnóstico se produjo después de tener descendencia, en un caso se le diagnosticó a la madre durante el primer embarazo.
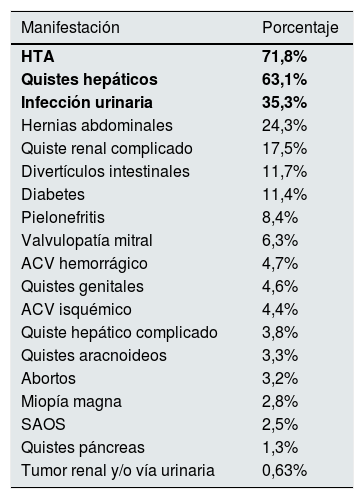
El principal motivo de diagnóstico son los antecedentes familiares con un 46,4% de los casos, seguido de HTA (8,9%), enfermedad renal crónica (8,6%), dolor abdominal-lumbar (7,4%), litiasis renal (6,8%) y hematuria (4,6%). De las manifestaciones extrarrenales, la más frecuente es la HTA (71,8%) seguida de los quistes hepáticos (63,1%). El resto de las manifestaciones extrarrenales se exponen en la tabla 3.
Manifestaciones extrarrenales y enfermedades asociadas
| Manifestación | Porcentaje |
|---|---|
| HTA | 71,8% |
| Quistes hepáticos | 63,1% |
| Infección urinaria | 35,3% |
| Hernias abdominales | 24,3% |
| Quiste renal complicado | 17,5% |
| Divertículos intestinales | 11,7% |
| Diabetes | 11,4% |
| Pielonefritis | 8,4% |
| Valvulopatía mitral | 6,3% |
| ACV hemorrágico | 4,7% |
| Quistes genitales | 4,6% |
| ACV isquémico | 4,4% |
| Quiste hepático complicado | 3,8% |
| Quistes aracnoideos | 3,3% |
| Abortos | 3,2% |
| Miopía magna | 2,8% |
| SAOS | 2,5% |
| Quistes páncreas | 1,3% |
| Tumor renal y/o vía urinaria | 0,63% |
En negrita, las tres manifestaciones extrarrenales más frecuentes.
De los 1.107 pacientes registrados, 642 están vivos (536 tienen su domicilio en Granada) y son atendidos en nuestro ámbito sanitario: el 70,2% en consultas externas, el 23,5% en trasplante renal, el 4,8% en HD y el 1,1% en DP. De ellos 246 (149 varones y 97 mujeres) están en una edad reproductiva adecuada y no tienen hijos aún el 50%.
La edad de inicio de TRS es 54,2±11,05 años: HD 54,6±11,20 años, DP 51,8±7,80 años y trasplante renal 52,2±8,50 años.
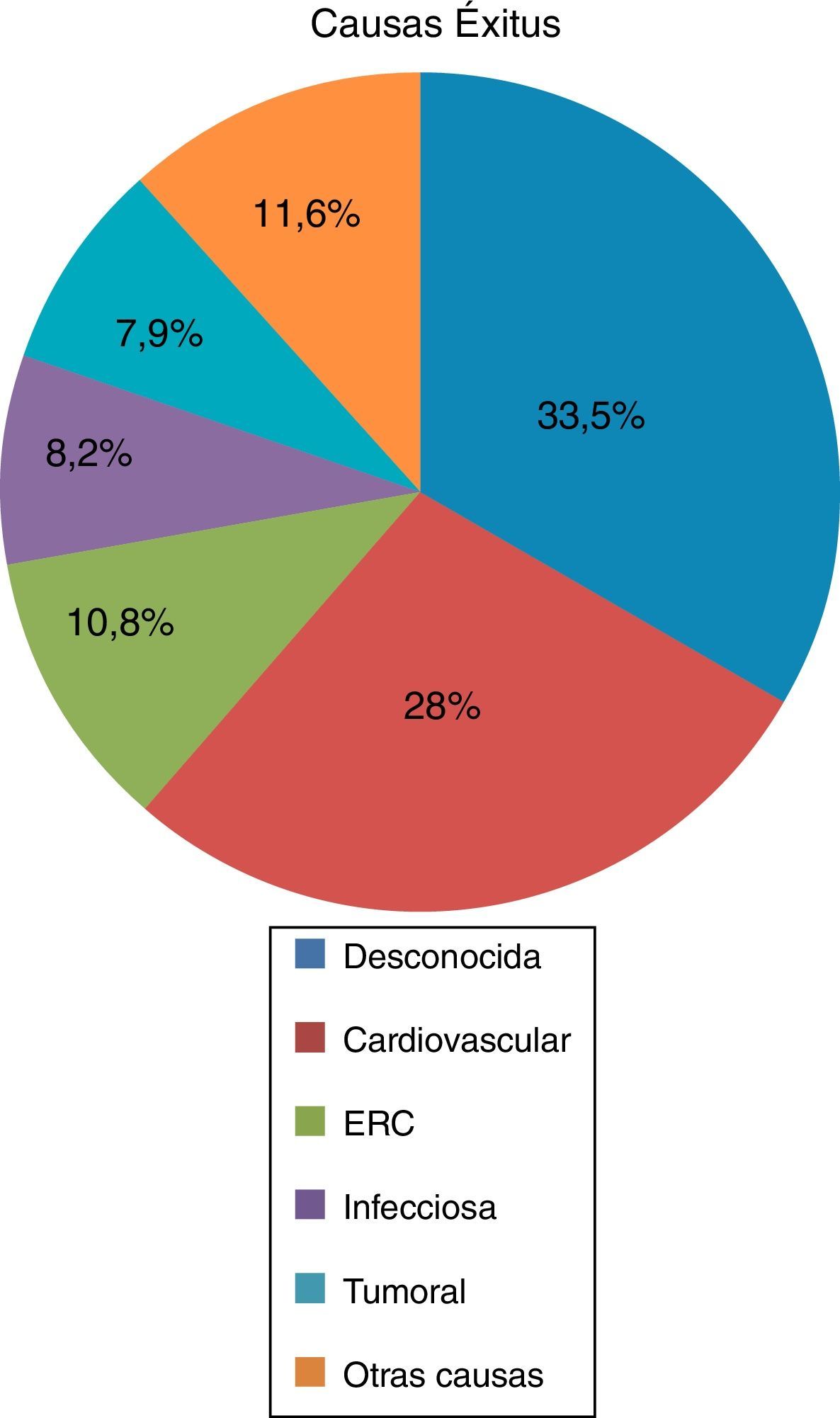
De los fallecidos (n=350), el 55,1% recibieron TRS en cualquiera de sus 3 modalidades, siendo de inicio la HD en el 53,9% seguida del trasplante renal con 17,6% de los casos y DP en un 4,6%. El 44,9% de los fallecidos no recibieron TRS, pero sin embargo, el 96,3% de los fallecidos tenían algún grado de insuficiencia renal en el momento del exitus. La edad media del exitus es de 60,97±14,10 años, siendo la principal causa de muerte la desconocida (33,5%) seguida de la cardiovascular (27,8%), insuficiencia renal (10,8%), infecciosa (8,2%), tumoral (7,9%), y otras causas en un 11,6% (fig. 2).

Hemos realizado estudio genético al menos a un miembro en 58 familias, cuyos resultados son el objetivo de otro trabajo. A modo de resumen, las mutaciones en PKD1 han sido las más frecuentes (84,5%), seguidas de las de PKD2 (6,9%) y PKD1&PKD2 (3,4%). En un 5,1% no hemos encontrado mutación detectable o eran variantes sin evidencia de ser patogénicas. Por el momento la mutación más llamativa y frecuente es c.10527_10528delGA ((p.Glu3509Aspfs*117) una deleción ubicada en el exón 35 del gen PKD1, que provoca un error de lectura y codón de parada, siendo la policistina 1 más corta. La presentan, por el momento, 6 familias (121 afectados conocidos), de la comarca de Loja, en la zona de Montefrío e Íllora, que no hemos podido emparentar mediante los árboles genealógicos. A esta deleción la denominamos mutación de Loja. De esta comarca aún tenemos varias familias sin estudio genético.
DiscusiónNuestro estudio surgió de la necesidad de conocer mejor la magnitud, forma de presentación y distribución de la enfermedad en nuestra región con el fin de atender adecuadamente las necesidades de la población.
La prevalencia de la PQRAD a día de hoy no está bien determinada y varía mucho de unos estudios a otros. Iglesias et al.1 establecen una prevalencia en los países occidentales de un caso/800-1.000 habitantes (entre 10-12,5 casos/10.000 habitantes) y sin embargo, estudios más recientes y en Europa establecen la prevalencia entre 5 y 10 casos/10.000 personas9–11. Según nuestros datos actuales, con 536 afectados vivos residentes en la provincia para una población de 915.392 habitantes (según los datos del Instituto Nacional de Estadística) en el área sanitaria de Granada la prevalencia estimada sería 1:1.707, o lo que es lo mismo, 5,85 casos/10.000 habitantes, pero mucho nos tememos que esta cifra está infraestimada. Entre las 251 familias con árbol genealógico realizado hay una media de 4,53 miembros no estudiados. Esto supone unas 1.155 personas: si descontamos los fallecidos, los que no viven en Granada y considerando una probabilidad de padecerla del 50%, habría aproximadamente unas 300 personas afectadas sin revisiones médicas. Si sumamos estos paciente a los registrados estaríamos en una cifra de 836 personas afectas, con la que tendríamos una prevalencia estimada de 1:1.094, cifra más acorde con lo descrito en la literatura.
Hemos demostrado que estamos frente a una enfermedad de distribución geográfica heterogénea; es decir, hay áreas donde se concentra más la enfermedad y otras en las que es prácticamente simbólica. El conocimiento de estas zonas nos permite desarrollar estrategias dirigidas hacia esas áreas y una distribución más eficiente de los recursos sanitarios.
El despistaje ecográfico rutinario a niños y adolescentes asintomáticos con algún padre afecto sigue siendo controvertido porque a día de hoy no hay un tratamiento efectivo y ya es conocido que un resultado ecográfico normal no excluye la enfermedad en las primeras etapas de la vida8. Sin embargo, creemos que es importante informar a las familias y ofrecer la posibilidad de realizar cribado ecográfico de la enfermedad en todos los miembros donde algún progenitor esté afecto, incluso en niños y adolescentes asintomáticos, por los siguientes motivos
- 1.
Realizar prevención primaria de la enfermedad, es decir, evitar la transmisión a las generaciones futuras de las personas afectas.
- 2.
Realizar prevención secundaria. Entendiendo por este término la puesta en marcha de todos los medios actuales para enlentecer la progresión de la enfermedad y disminuir el riesgo cardiovascular.
Según una encuesta realizada por la industria en el 201412, el 44% de los participantes españoles decidió no tener hijos tras el diagnóstico de la enfermedad. Y según otra encuesta realizada en el Reino Unido en el año 2014, el 59% de los pacientes con PQRAD de este país elegirían el diagnóstico genético preimplantacional (DGP) si estuviera disponible en la cartera de Servicios de la Seguridad Social Anglosajona13.
Un dato muy relevante es que la edad media del diagnóstico en nuestra provincia está en 34 años y en más de la mitad de los casos se produce después de haber tenido descendencia. Por lo tanto, debemos adelantarnos al diagnóstico si queremos realizar prevención primaria, y más ahora, desde que el 6 de noviembre de 2014 se publicara en el BOE la inclusión del DGP en aquellas enfermedades monogénicas sin posibilidad de tratamiento curativo y con diagnóstico genético posible, criterios que cumple la PQRAD14. Además, meses después, en julio de 2015 se incluyó también el DGP en la Cartera de Servicio de la Junta de Andalucía para enfermedades monogénicas susceptibles de dicho tratamiento15. El 29 de septiembre de 2016 fue aprobada en el Parlamento de Andalucía, la Proposición no de Ley para la creación del Plan de Prevención Primaria de la PQRAD a petición del GEEPAD y de la Asociación Amigos del Riñón con origen en Granada. En ella se insta a la Consejería de Sanidad a impulsar todas las medidas necesarias para que se lleven a cabo las técnicas reproductivas que eviten la transmisión de la enfermedad16.
Las Guías Clínicas Españolas de la PQRAD recomiendan que los pacientes afectos informen a sus familiares de primer grado sobre el riesgo de padecer la enfermedad y considera que debe ofrecerse despistaje de la misma así como consejo genético siempre17.
La tasa de mortalidad de los pacientes poliquísticos es casi 3 veces superior a la de la población general18. En nuestra región, la causa de exitus cardiovascular ha sido la segunda en frecuencia, otro motivo importante para un diagnóstico precoz. Actuando sobre los factores de riesgo cardiovascular mejoraremos las expectativas vitales y frenaremos la progresión de la insuficiencia renal de esta población.
Esta enfermedad debe tener un abordaje multidisciplinar entre las diversas especialidades intrahospitalarias y también con los profesionales de atención primaria. Son ellos los que están más cerca de las familias, los que pueden plantear el despistaje y el envío, en caso positivo, a las consultas más especializadas. Dentro del grupo multidisciplinar creemos que deben tener un papel importante los ginecólogos, los genetistas y los pediatras. Los ginecólogos, porque son los que van a aconsejar cuál es el método reproductivo más acorde con la situación de cada pareja, derivando a DGP cuando sea aconsejable. Los genetistas, porque son los que realizan e interpretan los estudios mutacionales y los que pueden realizar un adecuado consejo genético. Y los pediatras, que son los que deben estar vigilantes, para que los niños de padres con PQRAD puedan ser diagnosticados precozmente e identificar factores de riesgo que requieran de una intervención temprana19.
ConclusionesEl conocimiento de estos datos epidemiológicos sobre la PQRAD es fundamental para poder abordarla adecuadamente. En nuestra región la distribución de la PQRAD no es homogénea, y concentra focos donde la intervención sanitaria podría ser necesaria. Por otra parte, hemos observado que el diagnóstico se realiza de forma tardía y en más de la mitad de los casos después de tener descendencia. Si unimos esto a que además aún son numerosos los individuos sin diagnosticar, podemos concluir que es conveniente establecer un programa de información y despistaje interniveles donde el manejo a nivel asistencial del árbol genealógico es esencial.
La edad de exitus es inferior a la de la población general siendo la enfermedad cardiovascular la principal etiología; un diagnóstico precoz junto con un mayor control de los factores de riesgo cardiovascular podrían mejorar la esperanza de vida.
Nuestro grupo defiende la estrategia de prevención primaria basada en establecer el diagnóstico de la enfermedad antes de tener descendencia, el consejo genético e impulsando el empleo de técnicas reproductivas que eviten la transmisión de la enfermedad como el DGP, ya disponible en el sistema sanitario público.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A los enfermos y a las familias con PQRAD.



