




El conocimiento del papel del complemento en la patogenia del síndrome hemolítico urémico atípico y otras microangiopatías trombóticas (MAT) ha fomentado el desarrollo de la terapia anticomplemento con eculizumab más allá de su indicación original en la hemoglobinuria paroxística nocturna y en el síndrome hemolítico urémico atípico. La evidencia científica demuestra un estrecho límite entre MAT primarias y secundarias con activación del complemento subyacente en ambas. Por ello, el control del complemento se convierte en una diana terapéutica. El uso de eculizumab en MAT secundarias contempla 2escenarios: diagnóstico diferencial difícil entre MAT primaria y secundaria (incluidos cuadros clínicos incompletos) o daño por complemento en procesos distintos, donde se demuestra la eficacia del tratamiento. Esta revisión es una síntesis de la evidencia científica sobre el papel de la activación del complemento en la fisiopatología de las MAT secundarias y la eficacia de la terapia anticomplemento en MAT asociadas a embarazo, fármacos, trasplante, rechazo humoral, enfermedades sistémicas y glomerulonefritis. La experiencia es aún limitada, pero la respuesta a eculizumab en pacientes con MAT secundarias graves y refractarias al tratamiento convencional abre una puerta a la investigación de la terapia anticomplemento como nueva opción terapéutica.
Understanding the role of the complement system in the pathogenesis of atypical haemolytic uraemic syndrome and other thrombotic microangiopathies (TMA) has led to the use of anti-complement therapy with eculizumab in these diseases, in addition to its original use in patients with paroxysmal nocturnal haemoglobinuria andatypical haemolytic uraemic syndrome. Scientific evidence shows that both primary and secondary TMAs with underlying complement activation are closely related. For this reasons, control over the complement system is a therapeutic target. There are 2scenarios in which eculizumab is used in patients with TMA: primary or secondary TMA that is difficult to differentiate (including incomplete clinical presentations) and complement-mediated damage in various processes in which eculizumab proves to be efficacious. This review summarises the evidence on the role of the complement activation in the pathophysiology of secondary TMAs and the efficacy of anti-complement therapy in TMAs secondary to pregnancy, drugs, transplant, humoral rejection, systemic diseases and glomerulonephritis. Although experience is scarce, a good response to eculizumab has been reported in patients with severe secondary TMAs refractory to conventional treatment. Thus, the role of the anti-complement therapy as a new treatment option in these patients should be investigated.
La investigación del complemento en la patogenia del síndrome hemolítico urémico atípico (SHUa) y otras microangiopatías trombóticas (MAT) ha fomentado el desarrollo de la terapia anticomplemento. Eculizumab, primer anticuerpo monoclonal que bloquea el componente C5 del complemento, está aprobado para el tratamiento de la hemoglobinuria paroxística nocturna y el SHUa. Su eficacia en riñón nativo y en trasplante renal destacó la importancia de la activación del complemento en SHUa, prototipo de enfermedad por alteración primaria del complemento. Sin embargo, el diagnóstico y clasificación académica por exclusión de distintas etiologías e identificación de variantes genéticas pronto se complica ante la evidencia de situaciones en las que convergen distintas formas de MAT y la necesidad de diseñar categorías centradas en la patogenia y tratamiento.
Las primeras experiencias que amplían la indicación original de eculizumab en la hemoglobinuria paroxística nocturna y el SHUa parten de la evidencia científica del estrecho límite entre MAT primarias y secundarias1. Tres conceptos modifican la visión clásica de la MAT: la activación de complemento primaria o secundaria, el solapamiento entre distintas entidades clínicas y la disfunción endotelial como clave patogénica.
La MAT es un proceso complejo, derivado del desequilibrio entre inmunidad, coagulación y complemento, alterado por factores precipitantes (predominantes en MAT secundarias) en pacientes predispuestos por múltiples determinantes genéticos (dominantes en SHUa) (fig. 1).

La activación local (SHUa, isquemia/reperfusión) o sistémica del complemento, causa de lesión endotelial (rechazo humoral, glomerulonefritis C3 [GNC3], recidiva de nefropatía postrasplante) está presente en la MAT de forma primaria o secundaria. La demostración de la activación del complejo efector terminal (C5b-9) en MAT secundarias convierte al complemento en diana terapéutica y es la base del tratamiento con eculizumab2 en 2escenarios: situaciones de diagnóstico diferencial difícil entre MAT primaria y secundaria y entidades con activación de vía terminal de complemento con o sin trombosis microvascular:
- –
MAT de difícil diagnóstico:
- •
Desencadenada por infecciones (tabla 1)
Tabla 1.Pacientes con condiciones amplificadoras del complemento tratados con eculizumab
MAT secundaria Resultado Inducida por fármacos:
Doxorrubicina
Gemcitabina
Mitomicina
Mejoría de la función renal138
Mejoría de la función renal139
Remisión renal y hematológica140STEC-SHU
STEC-SHU y mutación en el gen CFHRemisión renal y neurológica141
Remisión hematológica, renal sin diálisis ni RP142PTT resistente a RP, vincristina, esteroides y rituximab Tratamiento con eculizumab, RP y esteroides143
Remisión hematológica, renal y neurológica143Deficiencia de cobalamina Eculizumab antes del diagnóstico144 PPT: púrpura trombótica trombocitopénica; RP: recambio plasmático; STEC-SHU: síndrome hemolítico urémico inducido por la infección de E. coli productora de toxina Shiga.
- •
Asociada a fármacos (tabla 1)
- •
Asociada a glomerulonefritis
- •
En enfermedades sistémicas (vasculitis, lupus, esclerodermia, etc.)
- •
En el embarazo y posparto
- •
Hipertensión arterial maligna (HTAM)
- •
De novo, postrasplante de órgano sólido (MAT-TOS).
- •
- –
Daño microvascular con activación de complemento:
- •
MAT en trasplante de progenitores hematopoyéticos (MAT-TPH)
- •
Rechazo mediado por anticuerpos (RMA)
- •
Daño por isquemia/reperfusión
- •
Púrpura trombótica trombocitopénica refractaria
- •
Nefropatía por anticuerpos antifosfolípidos.
- •
El paciente con MAT se identifica rápidamente en la práctica clínica, pero es difícil saber la causa. El tiempo que transcurre entre la sospecha diagnóstica y la certeza puede incidir negativamente en la eficacia del fármaco si la decisión terapéutica se demora. Los estudios genéticos y moleculares del complemento tienen valor pronóstico y estratégico a largo plazo, pero no para la indicación precoz, decisiva para preservar la función renal. La MAT es un proceso catastrófico y letal para el endotelio que conduce a daño renal y sistémico independientemente de su causa, con limitada respuesta a terapia convencional. Prueba de ello es que ensombrece el pronóstico y la supervivencia del paciente en todas las situaciones en las que complica la enfermedad de base: posparto, trasplante renal y TPH, HTAM, enfermedades autoinmunes o glomerulopatías.
El objetivo de esta revisión es aportar la evidencia científica actual sobre el tratamiento de las MAT secundarias y el manejo de estos pacientes desde una perspectiva terapéutica emergente y prometedora.
Microangiopatías trombóticas en enfermedades sistémicasExisten casos de MAT asociados a gran variedad de enfermedades sistémicas con solapamiento entre entidades y mutaciones del complemento hasta en un 33% de los pacientes con SHU asociado a enfermedades autoinmunes3,4. La presencia de diversos componentes del complemento en las biopsias renales plantea su papel patogénico, y la respuesta a eculizumab indica que la desregulación del complemento de base no genética puede desempeñar un papel importante y ser una posible diana terapéutica.
Vasculitis de anticuerpos anticitoplasma de neutrófilosEl concepto de vasculitis pauciinmunes está cambiando por el hallazgo de depósitos electrodensos en biopsia renal hasta en el 54% de los casos. En algunos pacientes con vasculitis de anticuerpos anticitoplasma de neutrófilos positiva se han encontrado componentes del complemento en los depósitos glomerulares (C3, C4, C1q, factor B, properdina y complejo de ataque de membrana) asociados con peor pronóstico renal5. La MAT en la biopsia renal asociada con vasculitis no es infrecuente (13,6%), especialmente en los casos graves y de peor curso evolutivo6. Recientemente, se ha demostrado que la activación de la vía alternativa del complemento tiene un papel primordial en la patogenia de las vasculitis7. Estudios experimentales con un antagonista del receptor C5a (CCX168) muestran un claro efecto beneficioso sobre la evolución de la afectación renal8. Por tanto, el uso de fármacos que bloqueen el complemento podría ser una alternativa terapéutica en estos pacientes.
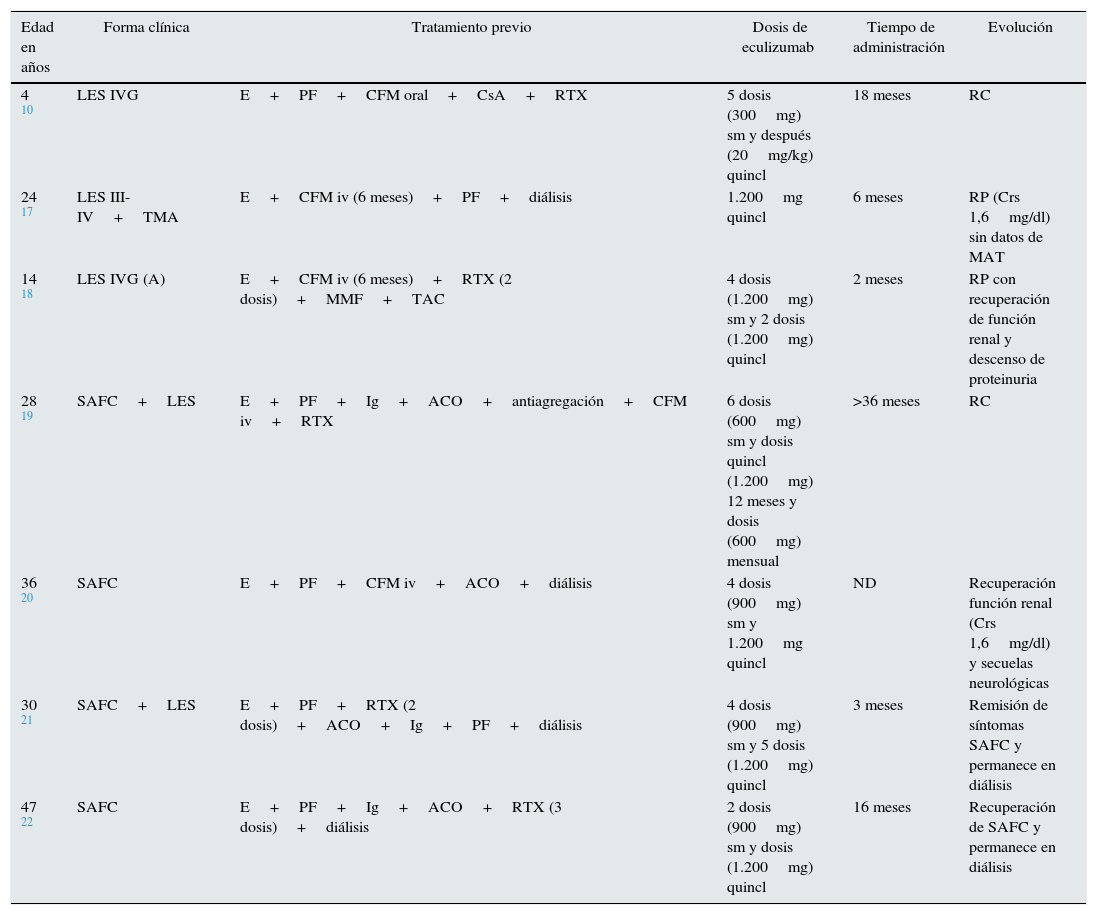
Lupus eritematoso sistémicoNumerosas observaciones clínicas señalan la importancia del complemento en la nefropatía lúpica (NL) y la traducción histológica es la lesión característica conocida como «full house», con depósito de inmunoglobulinas y complemento9 que juegan un doble papel en la patogenia de NL. Los componentes de la vía clásica (C1q, C2, C4) tienen un papel protector, facilitando la apoptosis de los inmunocomplejos del lupus eritematoso sistémico (LES), mientras que factores finales (C5 a C9) promueven inflamación y daño tisular a través de la generación de anafilotoxinas (C5a) y la formación del complejo de ataque de membrana (C5b-9)10. Un estudio experimental reveló el papel del déficit del factor H como potenciador del desarrollo de la NL con una presentación clínica e histológica más agresiva11. La coexistencia histológica de NL y MAT confiere peor pronóstico renal12. Song et al. encontraron MAT en el 24,3% de las biopsias renales con NL en un estudio retrospectivo. Estos pacientes tuvieron datos clínicos e histológicos más severos respecto al grupo sin MAT, lo que supone un factor de riesgo para la evolución de la función renal13. Estudios experimentales y clínicos han mostrado que la activación del complemento es esencial en la patogenia de la MAT y del LES, por lo que el uso de bloqueantes del complemento podría ser una terapia prometedora14. El tratamiento con eculizumab ha mostrado seguridad y buena tolerancia en estudios en fase i en pacientes con LES; desafortunadamente, no han continuado con estudios en fase ii o iii15,16. Sin embargo, 5 pacientes en la literatura, resistentes al tratamiento inmunosupresor habitual para NL (2 asociado a SAFC), responden positivamente al tratamiento con eculizumab (tabla 2)10,17,18.
Pacientes tratados con eculizumab en enfermedades sistémicas
| Edad en años | Forma clínica | Tratamiento previo | Dosis de eculizumab | Tiempo de administración | Evolución |
|---|---|---|---|---|---|
| 4 10 | LES IVG | E+PF+CFM oral+CsA+RTX | 5 dosis (300mg) sm y después (20mg/kg) quincl | 18 meses | RC |
| 24 17 | LES III-IV+TMA | E+CFM iv (6 meses)+PF+diálisis | 1.200mg quincl | 6 meses | RP (Crs 1,6mg/dl) sin datos de MAT |
| 14 18 | LES IVG (A) | E+CFM iv (6 meses)+RTX (2 dosis)+MMF+TAC | 4 dosis (1.200mg) sm y 2 dosis (1.200mg) quincl | 2 meses | RP con recuperación de función renal y descenso de proteinuria |
| 28 19 | SAFC+LES | E+PF+Ig+ACO+antiagregación+CFM iv+RTX | 6 dosis (600mg) sm y dosis quincl (1.200mg) 12 meses y dosis (600mg) mensual | >36 meses | RC |
| 36 20 | SAFC | E+PF+CFM iv+ACO+diálisis | 4 dosis (900mg) sm y 1.200mg quincl | ND | Recuperación función renal (Crs 1,6mg/dl) y secuelas neurológicas |
| 30 21 | SAFC+LES | E+PF+RTX (2 dosis)+ACO+Ig+PF+diálisis | 4 dosis (900mg) sm y 5 dosis (1.200mg) quincl | 3 meses | Remisión de síntomas SAFC y permanece en diálisis |
| 47 22 | SAFC | E+PF+Ig+ACO+RTX (3 dosis)+diálisis | 2 dosis (900mg) sm y dosis (1.200mg) quincl | 16 meses | Recuperación de SAFC y permanece en diálisis |
ACO: anticoagulación oral; CFM: ciclofosfamida; E: esteroides; Ig: inmunoglobulina; iv: intravenosa; LES: lupus eritematoso sistémico; MMF: micofenolato mofetil; ND: sin datos; PF: plasmaféresis; quincl: quincenales; RC: remisión completa; RP: remisión parcial; SAFC: síndrome antifosfolípido catastrófico; sm: semanal; RTX: rituximab; TAC: tacrolimus.
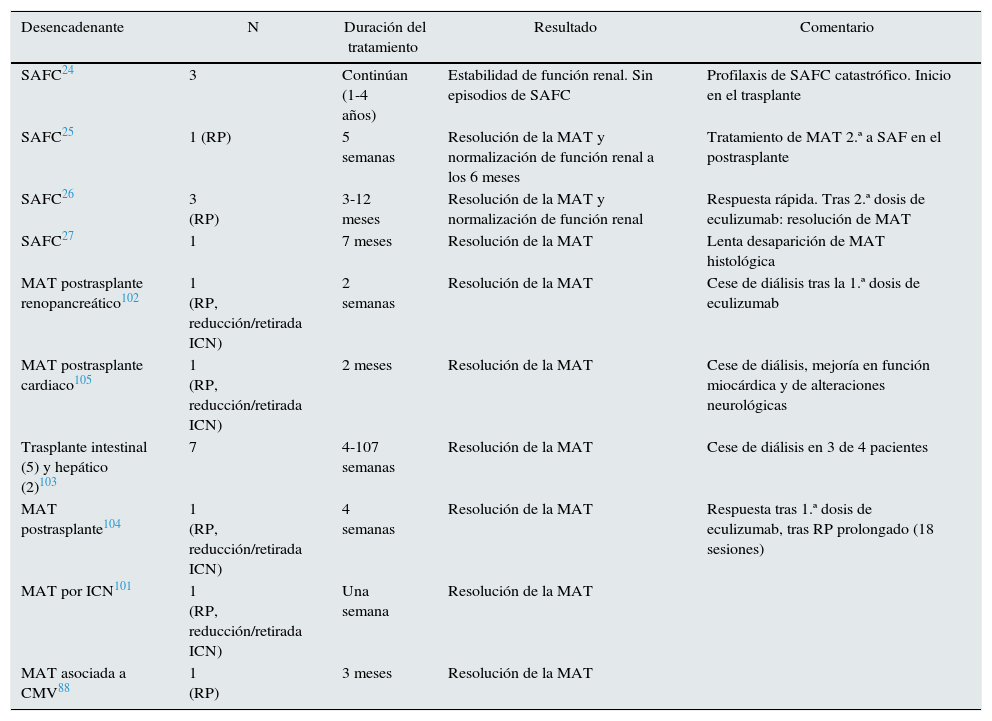
El síndrome antifosfolípido catastrófico (SAFC) es una variante del síndrome antifosfolípido (<1%) caracterizada por trombosis sistémica y desarrollo de un fracaso multiorgánico con elevada morbimortalidad y difícil tratamiento. Diversos autores han señalado que la activación incontrolada del complemento puede iniciar y amplificar los fenómenos característicos del SAFC, como la activación de endotelio, el factor de expresión de los monocitos y la agregación plaquetaria, unido a los hallazgos histológicos propios de la MAT19. El tratamiento abarca desde la anticoagulación hasta la terapia inmunosupresora (esteroides o ciclofosfamida), inmunoglobulinas y plasmaféresis. El uso de terapias que bloqueen el complemento puede ser una opción terapéutica, especialmente en pacientes refractarios al tratamiento habitual20. En la literatura 8 casos de SAFC, 4 trasplantados y 4 no trasplantados, han sido tratados exitosamente con eculizumab (tablas 2 y 3)19–27.
Utilización de eculizumab en MAT postrasplante de órgano sólido
| Desencadenante | N | Duración del tratamiento | Resultado | Comentario |
|---|---|---|---|---|
| SAFC24 | 3 | Continúan (1-4 años) | Estabilidad de función renal. Sin episodios de SAFC | Profilaxis de SAFC catastrófico. Inicio en el trasplante |
| SAFC25 | 1 (RP) | 5 semanas | Resolución de la MAT y normalización de función renal a los 6 meses | Tratamiento de MAT 2.ª a SAF en el postrasplante |
| SAFC26 | 3 (RP) | 3-12 meses | Resolución de la MAT y normalización de función renal | Respuesta rápida. Tras 2.ª dosis de eculizumab: resolución de MAT |
| SAFC27 | 1 | 7 meses | Resolución de la MAT | Lenta desaparición de MAT histológica |
| MAT postrasplante renopancreático102 | 1 (RP, reducción/retirada ICN) | 2 semanas | Resolución de la MAT | Cese de diálisis tras la 1.ª dosis de eculizumab |
| MAT postrasplante cardiaco105 | 1 (RP, reducción/retirada ICN) | 2 meses | Resolución de la MAT | Cese de diálisis, mejoría en función miocárdica y de alteraciones neurológicas |
| Trasplante intestinal (5) y hepático (2)103 | 7 | 4-107 semanas | Resolución de la MAT | Cese de diálisis en 3 de 4 pacientes |
| MAT postrasplante104 | 1 (RP, reducción/retirada ICN) | 4 semanas | Resolución de la MAT | Respuesta tras 1.ª dosis de eculizumab, tras RP prolongado (18 sesiones) |
| MAT por ICN101 | 1 (RP, reducción/retirada ICN) | Una semana | Resolución de la MAT | |
| MAT asociada a CMV88 | 1 (RP) | 3 meses | Resolución de la MAT |
CMV: citomegalovirus; ICN: inhibidor de la calcineurina; MAT: microangiopatía trombótica; RP: recambio plasmático; SAFC: síndrome antifosfolípido catastrófico.
La HTAM es un cuadro clínico caracterizado por marcada elevación de la presión arterial y retinopatía hipertensiva grado iii o iv. Recientemente se ha propuesto modificar esta definición por hipertensión severa con daño multiorgánico28. Esta entidad puede acompañarse de MAT con una frecuencia estimada del 5-20%29,30. Entre las hipótesis sobre la presencia de MAT en HTAM encontramos: hiperestimulación del sistema renina-angiotensina-aldosterona (valores elevados de aldosterona como principal factor humoral mediador de la MAT)31, factores genéticos como el genotipo TT de angiotensinógeno M235T y valores disminuidos del ADAMTS13 en pacientes con HTAM y MAT32,33. Por otro lado, hasta un 15% de los pacientes con MAT presentan HTAM. Es fundamental distinguir estas entidades, pues el tratamiento es diferente34. Algunos datos clínicos pueden orientar el diagnóstico diferencial a favor de una HTAM con MAT, como la historia previa de hipertensión, una presión arterial excesivamente elevada con deterioro de la función renal y trombocitopenia leve o persistente a pesar de controlar la presión arterial35.
Microangiopatías trombóticas en la glomerulonefritisLas glomerulopatías C3 (C3G) constituyen una entidad cuyas características clínicas, patogénicas y evolutivas han sido perfiladas en los últimos años. Su definición se basa en la presencia de depósitos intensos, aislados o claramente predominantes de C3 en la inmunofluorescencia36,37. Se distinguen 2tipos: GNC3 y enfermedad por depósitos densos; esta última, caracterizada por depósitos intensamente osmiofílicos, en forma de cinta, a lo largo de la membrana basal36. La patogenia de las C3G consiste en una activación anómala de la vía alternativa del complemento por mutaciones en los genes que codifican factores del complemento o proteínas reguladoras (factores H, I, CD46), o por autoanticuerpos contra estos factores reguladores36–38.
Diversos estudios han mostrado que el espectro de mutaciones genéticas y autoanticuerpos asociados a C3G es muy similar al de pacientes con SHUa36–38. Se postula que en C3G la desregulación del complemento se produce en fase fluida, lo que causa el acúmulo de los productos de la degradación del complemento en los capilares glomerulares, mientras que la activación del complemento en el SHUa afecta principalmente a superficies celulares (endotelio) y provoca una MAT grave36,38–40.
Los motivos por los que unos pacientes con determinada mutación genética presentan SHUa y otros C3G se conocen solo parcialmente39,40. No obstante, en la literatura se han descrito casos de SHUa/MAT y C3G como procesos coincidentes41 en un mismo paciente39,42–44, MAT en pacientes previamente diagnosticados de C3G42 y otros, que desarrollaron SHUa/MAT tras un trasplante renal43,44 y cuya causa de insuficiencia renal terminal había sido C3G.
El tratamiento de la C3G es controvertido. Aunque la inmunosupresión convencional había sido considerada inefectiva basándose en algunos casos clínicos y en series cortas de enfermos, un estudio reciente mostró un efecto favorable, sobre todo, de la basada en esteroides y micofenolato mofetilo (MMF)45. Datos preliminares de este estudio indican que los casos debidos a autoanticuerpos podrían ser especialmente sensibles al MMF, mientras que los causados por anomalías genéticas serían, en general, resistentes. Varios enfermos han sido tratados con eculizumab, con resultados diversos46–51, aunque el análisis cuidadoso de los casos señala que eculizumab podría ser efectivo en pacientes con enfermedad aguda y agresiva, con ausencia de lesiones crónicas avanzadas en la biopsia renal y con elevación de los niveles séricos de C5b-946–51. A día de hoy, no nos consta que se hayan descrito casos de pacientes con C3G que desarrollaran una MAT tratados con eculizumab.
Nefropatía por inmunoglobulina AEl sistema del complemento desempeña un papel destacado en la patogenia de la nefropatía por IgA (NIgA), amplificando el daño renal producido por el depósito de los inmunocomplejos compuestos por IgA1 deficiente en galactosa y sus autoanticuerpos específicos52–54. Determinados polimorfismos en genes del complemento influyen en la predisposición a sufrir NIgA52–54 y los depósitos de C4d y C3 tienen un significativo valor predictivo en esta entidad55.
La presencia de lesiones de MAT en biopsias renales de pacientes con NIgA ha sido señalada en algunos estudios56, pero necesita ser corroborada. Varios casos clínicos de MAT/SHUa y mutaciones en el factor H asociadas a NIgA han sido reportados57–59. Asimismo, se ha comunicado un efecto beneficioso de eculizumab en pacientes con NIgA agresiva sin MAT/SHUa concomitante60,61. Es evidente que se necesitan más estudios para determinar con precisión la incidencia real de MAT en la NIgA y la posible indicación terapéutica del bloqueo del complemento en esta entidad.
Otras glomerulonefritisLos intensos depósitos de diversos componentes del complemento observados en la mayoría de las glomerulonefritis evidencian que la activación del complemento juega un papel destacado en el daño glomerular de estos procesos. No existen estudios sistemáticos sobre la prevalencia de alteraciones genéticas o funcionales del complemento en las glomerulonefritis. Aparte de C3G e NIgA, se han descrito casos de MAT/SHUa en pacientes con glomeruloesclerosis segmentaria y focal, nefropatía membranosa, aguda postinfecciosa y membranoproliferativa por inmunocomplejos62. No existen casos tratados con eculizumab en estas MAT asociadas a glomerulonefritis, aunque ocasionalmente este fármaco ha sido usado con buenos resultados en glomerulonefritis mediadas por inmunocomplejos63.
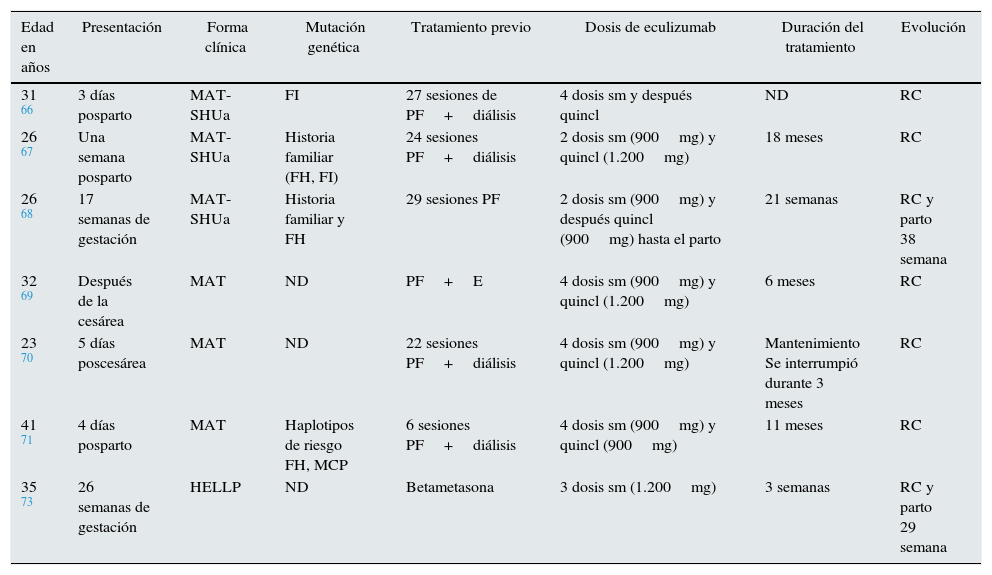
Microangiopatías trombóticas en el embarazoLa MAT asociada al embarazo es una entidad clínica infrecuente con incidencia estimada de un caso por cada 25.000 embarazos y elevada morbimortalidad maternal y perinatal. Durante la última década, 2hallazgos han permitido mejorar el conocimiento de esta entidad: la deficiencia adquirida o congénita del ADAMTS13, causante de la púrpura trombótica trombocitopénica del segundo y tercer trimestres del embarazo, y el desequilibrio de los factores reguladores de la vía alternativa del complemento como factor genético de riesgo para el desarrollo de SHUa posparto. Fakhouri et al. describieron que un 85% de las pacientes con SHU asociado al embarazo presentaban alteraciones en los factores del complemento64. Aunque el mecanismo patogénico está todavía por dilucidar, se plantea que, en un embarazo normal con alguna mutación en los genes del complemento, tendría lugar una activación descontrolada contrarrestada por proteínas reguladoras situadas en la superficie del trofoblasto como CD55 o DAF (factor acelerador de decaimiento), MCP y CD59. Sin embargo, numerosos desencadenantes (fenómenos inflamatorios, infecciones, hemorragias) podrían reactivar la vía alternativa del complemento después del parto y, en ausencia de mecanismos regulatorios presentes en la superficie placentaria, precipitarían la MAT posparto65. La implicación del complemento en la MAT del embarazo ha permitido el uso de eculizumab en 7 pacientes, con o sin mutación del complemento, en embarazo o posparto, con diferentes pautas, pero en todos los casos con una excelente respuesta clínica (tabla 4)66–71.
Pacientes tratados con eculizumab en casos de MAT asociada al embarazo
| Edad en años | Presentación | Forma clínica | Mutación genética | Tratamiento previo | Dosis de eculizumab | Duración del tratamiento | Evolución |
|---|---|---|---|---|---|---|---|
| 31 66 | 3 días posparto | MAT-SHUa | FI | 27 sesiones de PF+diálisis | 4 dosis sm y después quincl | ND | RC |
| 26 67 | Una semana posparto | MAT-SHUa | Historia familiar (FH, FI) | 24 sesiones PF+diálisis | 2 dosis sm (900mg) y quincl (1.200mg) | 18 meses | RC |
| 26 68 | 17 semanas de gestación | MAT-SHUa | Historia familiar y FH | 29 sesiones PF | 2 dosis sm (900mg) y después quincl (900mg) hasta el parto | 21 semanas | RC y parto 38 semana |
| 32 69 | Después de la cesárea | MAT | ND | PF+E | 4 dosis sm (900mg) y quincl (1.200mg) | 6 meses | RC |
| 23 70 | 5 días poscesárea | MAT | ND | 22 sesiones PF+diálisis | 4 dosis sm (900mg) y quincl (1.200mg) | Mantenimiento Se interrumpió durante 3 meses | RC |
| 41 71 | 4 días posparto | MAT | Haplotipos de riesgo FH, MCP | 6 sesiones PF+diálisis | 4 dosis sm (900mg) y quincl (900mg) | 11 meses | RC |
| 35 73 | 26 semanas de gestación | HELLP | ND | Betametasona | 3 dosis sm (1.200mg) | 3 semanas | RC y parto 29 semana |
E: esteroides; FI: factor I; FH: factor H; MAT: microangiopatía trombótica; ND: no datos; PF: plasmaféresis; quincl: quincenales; RC: remisión completa; SHUa: síndrome hemolítico urémico atípico; sm: semanales.
Recientemente esta activación del complemento también se ha asociado a otras entidades relacionadas con el embarazo como la preeclampsia y el síndrome de anemia hemolítica, trombocitopenia y elevación de las enzimas hepáticas (síndrome HELLP). La preeclampsia representa la respuesta maternal a un exceso de factores antiangiogénicos generados por la hipoperfusión placentaria que incluyen factores vasculopáticos como SolubleFms-Like Tyrosine kinase 1 (sFlt-1), potente antagonista del vascular endothelial growth factor (VEGF) y la endoglina (inhibidor del TGF-B). La clínicas varían desde diferentes grados de hipertensión hasta un síndrome HELLP o eclampsia65,72. El sistema del complemento es clave en los procesos inflamatorios y su sobreactivación induce la desregulación de los factores angiogénicos que contribuyen en la patogenia. Basándose en este argumento, Burwick et al. presentaron el caso de una mujer con síndrome HELLP grave a las 26 semanas de gestación, tratada con eculizumab, con respuesta clínica favorable que permitió prolongar el embarazo 17 días y disminuir la morbimortalidad del neonato73.
El complemento en el trasplante de órgano sólidoMicroangiopatías trombóticas postrasplante renal y de otros órganos sólidosLa MAT-TOS es una complicación poco frecuente observada en entre un 0,8 y un 15% de los trasplantes renales. Suele aparecer en los primeros 3 meses (2/3 de los pacientes) y conlleva pérdida de injerto hasta en 1/3 de ellos74–77.
Clínica y patológicamente, la MAT-TOS es indistinguible del SHUa recurrente76. Los antecedentes personales o familiares, el inicio abrupto y la mayor afectación sistémica y hematológica son más frecuentes en el SHUa recurrente3. La MAT-TOS tiene una presentación variable, desde proteinuria e hipertensión aisladas (30% de casos) hasta MAT completa (anemia hemolítica microangiopática, trombocitopenia y deterioro de la función renal) con mayor riesgo de pérdida del injerto74,76,78.
El SHUa se relaciona con mutaciones en proteínas reguladoras del complemento. En cambio, en MAT-TOS intervienen múltiples factores de daño endotelial y activación de complemento, como donantes con criterios expandidos, fenómenos asociados a muerte cerebral, isquemia/reperfusión, infecciones virales, rechazo humoral, anticuerpos antifosfolípidos, anticuerpos anticardiolipina y anti-VHC e inmunosupresores, sobre todo inhibidores de la calcineurina (ICN) e inhibidores de mTOR (imTOR)26. Por otro lado, se observan variantes genéticas de proteínas del complemento hasta en el 30% de los pacientes con MAT-TOS74.
En la muerte cerebral y en la isquemia/reperfusión, aumenta la liberación de C5a y C5b-9 y disminuye la capacidad de unión de CFH al endotelio26,78–83, lo que favorece la lesión endotelial más marcada observada en órganos de donantes en parada cardiorrespiratoria84. En modelos experimentales se ha observado reducción del daño tisular al bloquear el complemento84,85. Actualmente, se lleva a cabo un ensayo clínico para evaluar el efecto de eculizumab sobre la función retrasada del injerto (NCT02145182).
Las infecciones virales pueden desencadenar MAT-TOS y recurrencia de SHUa86–89. Aunque la patogenia es desconocida, está relacionada con el trofismo endotelial del virus que induce expresión de moléculas de adhesión y liberación del factor von Willebrand, que causa adhesión plaquetaria y trombosis microvascular88. Hasta el momento se han descrito 7 casos en la literatura por citomegalovirus postrasplante renal86–89. En todos, la administración de ganciclovir intravenoso y recambios plasmáticos logró resolver la hemólisis. Una publicación reciente ha mostrado una segunda recurrencia de MAT asociada a viremia por citomegalovirus resuelta tras tratamiento con valganciclovir, que facilitó el aclaramiento de la viremia, más eculizumab, que puede prevenir la destrucción celular producida por el citomegalovirus y mediada por el complemento88.
El tratamiento inmunosupresor es uno de los principales factores de riesgo para MAT-TOS. Se observa en entre el 4 y el 15% de los pacientes tratados con ciclosporina y el 1-4% de los tratados con tacrolimus75,79,90. Los ICN tienen un efecto tóxico endotelial directo mediado por la formación de micropartículas que estimulan la vía alterna de C y por isquemia tisular (reducción de vasodilatadores prostaciclina y óxido nítrico), formación de especies reactivas de O2 y aumento de agregación plaquetaria90–97. Los imTOR favorecen la MAT postrasplante por inhibición de la síntesis de VEGF por los podocitos, que favorece el daño endotelial, y la reducción de la capacidad de regeneración del endotelio. El riesgo de MAT-TOS se incrementa de forma significativa al asociar ambos fármacos92,98–100.
El manejo de la MAT-TOS no está bien establecido. Inicialmente se recomienda la eliminación del agente causal (tratamiento antiviral, del rechazo, etc.). En caso de MAT por fármacos, reducir o retirar el inmunosupresor relacionado puede resolver la MAT, pero aumenta el riesgo de rechazo agudo. Para minimizar este riesgo, el cambio a belatacept podría mantener la inmunosupresión sin aumentar el riesgo de MAT100. Si persiste la MAT o en casos graves, se recomienda asociar recambio plasmático. Pese a ello, el riesgo de pérdida de injerto continúa siendo del 20-42%74,77,78. Dada la implicación del sistema del complemento en la MAT postrasplante y su mal pronóstico, en casos refractarios a terapia habitual se ha utilizado terapia anticomplemento en diferentes órganos (tabla 3)24,25,27,75,101–105. En una revisión retrospectiva, Dhakal describe una tasa de recuperación hematológica y mejoría de la función renal en el 90% de 26 casos con MAT-TPH y TOS tratados con eculizumab. Aunque la dosis, frecuencia y duración del tratamiento es variable (900 y 1.200mg/dosis entre 2 y 21 dosis), el promedio de respuesta se observa tras 2 dosis (entre 1 y 18)75.
Con un limitado número de casos heterogéneos, estas evidencias apoyan que eculizumab puede ser una opción terapéutica en MAT de novo post-TOS, aunque son necesarios más estudios que indiquen qué grupo de pacientes se beneficiarían, qué dosis y cuál es la duración óptima del tratamiento.
Rechazo mediado por anticuerposEl RMA es un problema mayor del trasplante renal en pacientes con anticuerpos human leukocyte antigen (HLA) pre- y postrasplante, debido al riesgo de pérdida del injerto, rápido deterioro de función y resistencia al tratamiento. Se presenta en entre el 30 y el 40% de los pacientes sensibilizados a pesar de inmunosupresión, eliminación de anticuerpos y esplenectomía.
Eculizumab se ha utilizado en prevención y tratamiento del RMA por la evidencia del papel patogénico crítico del complemento. Los anticuerpos específicos contra donante activan el complemento sobre el endotelio, desencadenando el rechazo agudo y facilitando la inflamación en el rechazo crónico. La isquemia/reperfusión y los inmunosupresores contribuyen a la amplificación del complemento y a la pérdida de resistencia endotelial a la respuesta inmune y la trombosis.
La eficacia de eculizumab en RMA no está asociada a MAT histológica. Aunque esta se encuentra en entre el 4 y el 46% de los pacientes, eculizumab no solo incrementa el dintel para el desarrollo de MAT, sino que interfiere en la respuesta inmune. El bloqueo específico C5 previene la formación de anafilotoxina C5a (potente inductor de respuesta inflamatoria) y del complejo de ataque de membrana C5b-9, que daña directamente el endotelio1.
La mayor evidencia del poder del control de complemento es el fenómeno de acomodación: protección adquirida del injerto con C4d positivo e histología normal. En el trasplante ABO incompatible por anticuerpos anti-A/B, aumenta la expresión de proteínas reguladoras CD55 y CD59. Aunque utilizado precozmente postrasplante eculizumab previene el rechazo agudo, no existe evidencia de acomodación en pacientes tratados2.
Eculizumab en prevención de rechazo mediado por anticuerposLa experiencia más extensa ha sido publicada por Stegall et al.106. Veintiséis receptores de trasplante de donante vivo sensibilizados recibieron eculizumab para prevenir RMA. Comparados con 51 pacientes históricos no tratados, la incidencia de RMA fue del 7,7% en el grupo tratado frente al 40%. Dos pacientes con altos niveles de anticuerpos donante específicos (DSA) presentaron RMA durante el tratamiento.
La respuesta a eculizumab varía en distintos tipos de rechazo. Bentall107 identificó DSA de tipo IgM en 4 casos de la cohorte de Stegall, 2 de ellos con RMA y uno con RMA subclínico. Burbach et al. publicaron el caso de 2pacientes con mala evolución en rechazo C4d negativo108.
El desarrollo de DSA IgM, daño endotelial directo por anticuerpos, citotoxicidad celular anticuerpos-dependiente/complemento-independiente, activación de la vía alternativa o lectinas y lesión inflamatoria por componentes de complemento proximal podrían ser mecanismos relacionados con la falta de respuesta a eculizumab.
Eculizumab en el tratamiento del rechazo mediado por anticuerposLa literatura incluye una serie retrospectiva con 10 pacientes y 10 casos independientes109. Orandi et al. trataron con eculizumab (con o sin esplenectomía) 10 de 24 casos con RMA grave110. Al año se perdieron 4 de 5 casos tratados con eculizumab; ninguno de los 5 tratados con eculizumab más esplenectomía. Ocho de otros 10 casos independientes respondieron al tratamiento y 2 perdieron el injerto (fallo a 47 días, nefropatía BK). Un caso pediátrico publicado recientemente subraya la importancia del tratamiento temprano: tratado precozmente con 2dosis de eculizumab mejoró la función renal y normalizó la histología111.
La patogenia del RMA es compleja. Eculizumab no influye sobre los niveles de DSA circulantes ni sobre la respuesta celular independiente de complemento, por lo que debe ser combinado con timoglobulina, rituximab, recambio plasmático e IGIV. En la cohorte de Stegall et al., la duración del tratamiento fue variable (3-4 meses). En este sentido es importante el nivel de DSA y se propone mantener tratamiento con MFI>9.000112.
Eculizumab ha mostrado eficacia en la prevención del RMA en pacientes sensibilizados y en el tratamiento del RMA refractario. La repercusión sobre la glomerulopatía del trasplante es difícil de valorar por las diferencias en la gravedad del RMA y los tratamientos asociados. El inicio precoz se relaciona con mayor eficacia y es necesario aumentar la experiencia para establecer la duración óptima del tratamiento en relación con la intensidad de DSA.
Microangiopatía trombótica asociada al trasplante de progenitores hematopoyéticosLa MAT-TPH es una complicación con mortalidad elevada (hasta el 90% en pacientes graves92) y con riesgo de nefropatía crónica en los menos graves. Se ha estimado que la MAT ocurre en entre el 10 y el 35% de los TPH, especialmente después del trasplante alogénico113–117. Entre los factores desencadenantes destacan los ICN para la profilaxis de la enfermedad del injerto contra el huésped, que causan directamente lesión endotelial, activan el complemento y alteraran la actividad de ADAMTS13 (tabla 5)97. Aunque la enfermedad del injerto contra el huésped aguda es un factor de riesgo independiente de MAT-TPH, no se ha establecido relación causal118–120.
Factores de riesgo de MAT-TPH
| Factores de riesgo115,145–150 |
|---|
| Edad avanzada |
| EICH |
| Acondicionamiento con radioterapia |
| Sexo femenino |
| Inhibidores de la calcineurina |
| Sirolimus |
| Disparidad HLA |
EICH: enfermedad del injerto contra el huésped; HLA: human leukocyte antigen.
La MAT-TPH puede tener un curso benigno sin tratamiento o con reducción de la dosis del ICN. Sin embargo, algunos pacientes desarrollan una lesión sistémica con daño renal, gastrointestinal, serositis, hipertensión pulmonar y fallo multiorgánico121–124. La lesión endotelial más común es la renal, que provoca descenso del filtrado glomerular, proteinuria e hipertensión arterial. La afectación pulmonar provoca hipoxemia o distrés respiratorio125,126 y la gastrointestinal, vómitos, diarrea, dolor abdominal y hemorragia, que se solapan con el propio cuadro de la enfermedad del injerto contra el huésped intestinal, y que precisa estudio histológico para el diagnóstico diferencial.
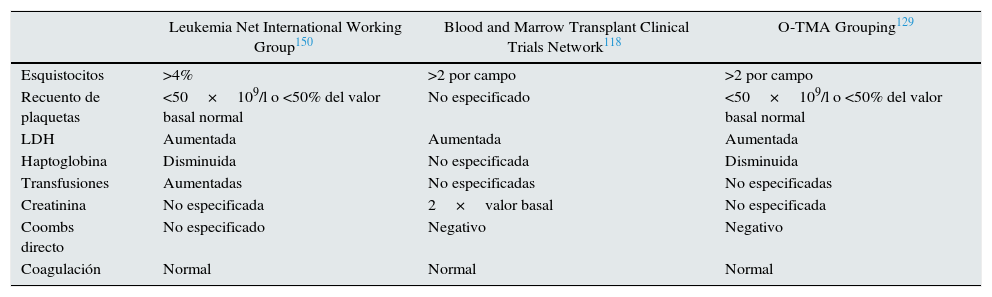
La MAT-TPH debe sospecharse ante elevación brusca de lactato deshidrogenasa sérica (LDH), hipertensión y proteinuria. El aumento de LDH es secundario al daño tisular por isquemia relacionada con trombosis y lesión endotelial127. Los esquistocitos suelen aparecer tardíamente. La tabla 6 recoge los criterios diagnósticos de MAT-TPH.
Comparación de los criterios diagnósticos de MAT-TPH
| Leukemia Net International Working Group150 | Blood and Marrow Transplant Clinical Trials Network118 | O-TMA Grouping129 | |
|---|---|---|---|
| Esquistocitos | >4% | >2 por campo | >2 por campo |
| Recuento de plaquetas | <50×109/l o <50% del valor basal normal | No especificado | <50×109/l o <50% del valor basal normal |
| LDH | Aumentada | Aumentada | Aumentada |
| Haptoglobina | Disminuida | No especificada | Disminuida |
| Transfusiones | Aumentadas | No especificadas | No especificadas |
| Creatinina | No especificada | 2×valor basal | No especificada |
| Coombs directo | No especificado | Negativo | Negativo |
| Coagulación | Normal | Normal | Normal |
LDH: lactato deshidrogenasa; O-TMA: overall thrombotic microangiopathy.
La determinación de la actividad del complemento terminal mediante la cuantificación de niveles plasmáticos del complejo efector terminal (sC5b-9) permitiría identificar a los pacientes que pueden beneficiarse de una terapia anticomplemento. Niveles plasmáticos elevados del sC5b-9 y proteinuria se han asociado con una reducción de la tasa de supervivencia (<20% al año)128.
Los niveles plasmáticos de ADAMTS13, habitualmente moderadamente disminuidos, permiten excluir con seguridad la púrpura trombótica trombocitopénica, extremadamente rara tras el TPH.
Por último, en receptores de trasplante alogénico, el estudio genético del sistema del complemento debe realizarse en muestras no hematológicas, como frotis bucal.
El tratamiento inicial consiste en la retirada de agentes potencialmente desencadenantes (ICN), control de complicaciones asociadas (infecciones, enfermedad de injerto contra huésped) y adecuada terapia antihipertensiva. Sin embargo, la respuesta es a menudo limitada, especialmente en pacientes con MAT-TPH grave. Las opciones terapéuticas actuales incluyen recambio plasmático, defibrótido, rituximab y eculizumab.
El recambio plasmático está debatido y se asocia a una tasa de respuesta del 36% (entre 0 y 80%) probablemente porque se reserva para casos graves. La mejoría de los parámetros hematológicos (plaquetas, hemoglobina, haptoglobina, LDH) puede dar falsa impresión de mejoría del trastorno subyacente. Las proteínas reguladoras del complemento del plasma infundido pueden conseguir remisión a corto plazo, pero el daño tisular y la mortalidad no se modifican129,130. Si se indica recambio plasmático, deberá comenzarse precozmente, con frecuencia diaria hasta la resolución de la MAT.
El anticoagulante defibrótido, aprobado en Europa, se ha usado en pacientes con manifestaciones leves y la administración de rituximab solo o combinado con otros agentes también se ha asociado a respuesta favorable en casos seleccionados131.
La MAT-TPH es una enfermedad multifactorial en la que se activa el complemento por vía clásica y alternativa, que resulta en daño tisular por trombosis microvascular132. Existen evidencias crecientes del desequilibrio del complemento implicado en algunos casos de MAT-TPH y de remisión completa con terapia anticomplemento con eculizumab. Se ha comprobado que los pacientes con MAT-TPH tienen activación del complemento, autoanticuerpos antifactor H y depósito renal C4d133–135 y pueden presentar variantes patogénicas de genes de complemento de SHUa. En un estudio pediátrico de 6 niños con fracaso renal agudo y MAT-TPH, la mayoría presentaban deleción de genes de proteínas relacionadas con el factor H (CFHR3 y CFHR1), 3de ellos con autoanticuerpos anti-FH. La respuesta al recambio plasmático fue mala y se detectaron niveles elevados de sC5b-C9 y trombosis en biopsia renal. De los 6 pacientes, 4 alcanzaron niveles plasmáticos terapéuticos de eculizumab y respuesta clínica136.
En un estudio retrospectivo,12 pacientes con MAT-TPH grave recibieron eculizumab en Francia entre 2010 y 2013, con un seguimiento mediano de 14 meses: la respuesta hematológica y global fue del 50 y del 33%, respectivamente137.
Tanto las dosis de eculizumab como el tiempo de respuesta para controlar la MAT-TPH son mayores que las observadas habitualmente en el SHUa, y se recomienda evaluar la respuesta tras una inducción al menos de 4-6 semanas.
DiscusiónLa implicación del complemento en el SHUa y la MAT de distintas etiologías descubre una nueva perspectiva terapéutica: el control del complemento para evitar el daño endotelial e inflamatorio. La experiencia muestra que el espectro de la MAT no está formado por categorías disyuntivas sino que existe un contínuum en la patogenia donde intervienen 2elementos que interactúan con intensidad variable: factores genéticos de predisposición y factores etiológicos o predisponentes. La evidencia científica muestra solapamiento y mecanismos comunes entre SHUa y otras MAT secundarias en las que se produce daño endotelial por el complemento, hecho que explica la respuesta a eculizumab en pacientes con MAT de distinta etiología y lo convierte en una terapia prometedora. La contribución del complemento en la patogenia de las MAT secundarias está bien documentada en las entidades revisadas en este artículo. Son necesarios estudios sistemáticos que analicen las variantes genéticas y de predisposición a la activación de las vías alternativa y terminal, las consecuencias funcionales de esta activación sobre los mecanismos de la enfermedad y el efecto funcional/clínico del bloqueo del componente C5 por eculizumab, que en MAT secundarias representa una terapia coadyuvante al tratamiento convencional de la enfermedad de base. La falta de respuesta al control del factor etiológico y al tratamiento convencional en MAT secundarias ha impulsado la terapia con eculizumab en la mayoría de los casos publicados.
Las limitaciones fundamentales para evaluar la eficacia de eculizumab en MAT secundarias son la heterogeneidad clínica y patogénica de los casos generalmente graves y refractarios, el sesgo de publicación, la concurrencia de otras terapias utilizadas en la enfermedad de base, la variación en las dosis, frecuencia y duración de los tratamientos y la dificultad de la monitorización con marcadores biológicos y farmacocinéticos. La duración óptima del tratamiento es una cuestión importante, dado el impacto económico de eculizumab. Este es además un factor limitante del tratamiento precoz que, como se ha demostrado en el SHUa, es más coste-efectivo. Las pautas de administración en la mayoría de los casos corresponden a la indicación en SHUa y hemoglobinuria paroxística nocturna o están modificadas de forma empírica.
Aunque el número global de pacientes tratados todavía no es elevado, actualmente se dispone de un número significativo de casos tratados exitosamente con eculizumab en MAT postembarazo/parto, post-TOS, TPH y en prevención/tratamiento del RMA. Los casos de MAT en enfermedades sistémicas, asociada a fármacos, glomerulonefritis e HTAM constituyen el grupo más heterogéneo y difícil de evaluar.
La implicación de la activación de complemento en los mecanismos patogénicos de las MAT secundarias es clara, pero es necesario impulsar estudios para sistematizar la experiencia clínica y permitir el diseño de estrategias comunes en distintas enfermedades y la identificación de marcadores biológicos y de parámetros clínicos de eficacia que permitan determinar con precisión la posible indicación terapéutica en cada caso.
Conceptos clave- •
La MAT es un proceso complejo, derivado del desequilibrio entre inmunidad, coagulación y complemento por una combinación de factores etiológicos (MAT secundarias) y factores de riesgo genético (dominantes en SHUa).
- •
La clasificación de MAT clásica no explica la complejidad de los mecanismos de la enfermedad ni refleja los objetivos terapéuticos, por lo que es necesario reconsiderar una clasificación patogénica de la MAT.
- •
Existe solapamiento entre ambas entidades y mecanismos comunes de daño endotelial mediado por el complemento que explican la respuesta a eculizumab en pacientes con MAT de distinta etiología, lo que lo convierte en una terapia prometedora.
- •
Aunque la experiencia es limitada, eculizumab se ha mostrado eficaz en la MAT post-TOS, TPH, prevención y tratamiento de rechazo humoral, asociadas a embarazo y enfermedades sistémicas. La precocidad del tratamiento, al igual que en SHUa, se asocia con mejor beneficio terapéutico.
- •
Son necesarios más estudios para determinar con precisión la indicación, dosificación y duración del tratamiento como terapia coadyuvante al tratamiento etiológico en cada caso.
Elena Román, Javier de la Rubia, Amparo Sempere, Enrique Morales, Manuel Praga y Ana Ávila han desarrollado actividades de consultoría y docencia para Alexion Pharmaceuticals. Elena Román, Enrique Morales y Manuel Praga han formado parte de comités de expertos en SHUa. Ninguna de las actividades mencionadas ha influido en la elaboración de este manuscrito.
Elena Román, Santiago Mendizábal, Isidro Jarque, Ana Ávila y José Luis Górriz son miembros de Medicamentos de Alto Impacto Sanitario o Económico (MAISE) para eculizumab, de la Conselleria de Sanidad Universal y Salud Pública de la Generalitat Valenciana.
Los autores desean agradecer a Alexion Pharmaceuticals el soporte logístico para la realización de la reunión.



