
Presentamos el caso de una niña de 22 meses de edad, evaluada de forma multidisciplinaria por presentar crisis convulsivas de difícil manejo que inició a los 6 meses y poliquistosis renal bilateral. Los padres son sanos y no consanguíneos. Es producto de una segunda gestación, embarazo simple no controlado, complicado con síndrome de HELP. Obtenida por cesárea segmentaria a las 36 semanas, requirió reanimación al momento del nacimiento y presentó cianosis generalizada. El peso al nacer fue de 1.700g (DE −3,8) y la talla 46cm (DE −2,5). Fue hospitalizada a los 8 meses por crisis convulsivas tónico-clónicas, el cual ha presentado evolución tórpida y retraso global del desarrollo.
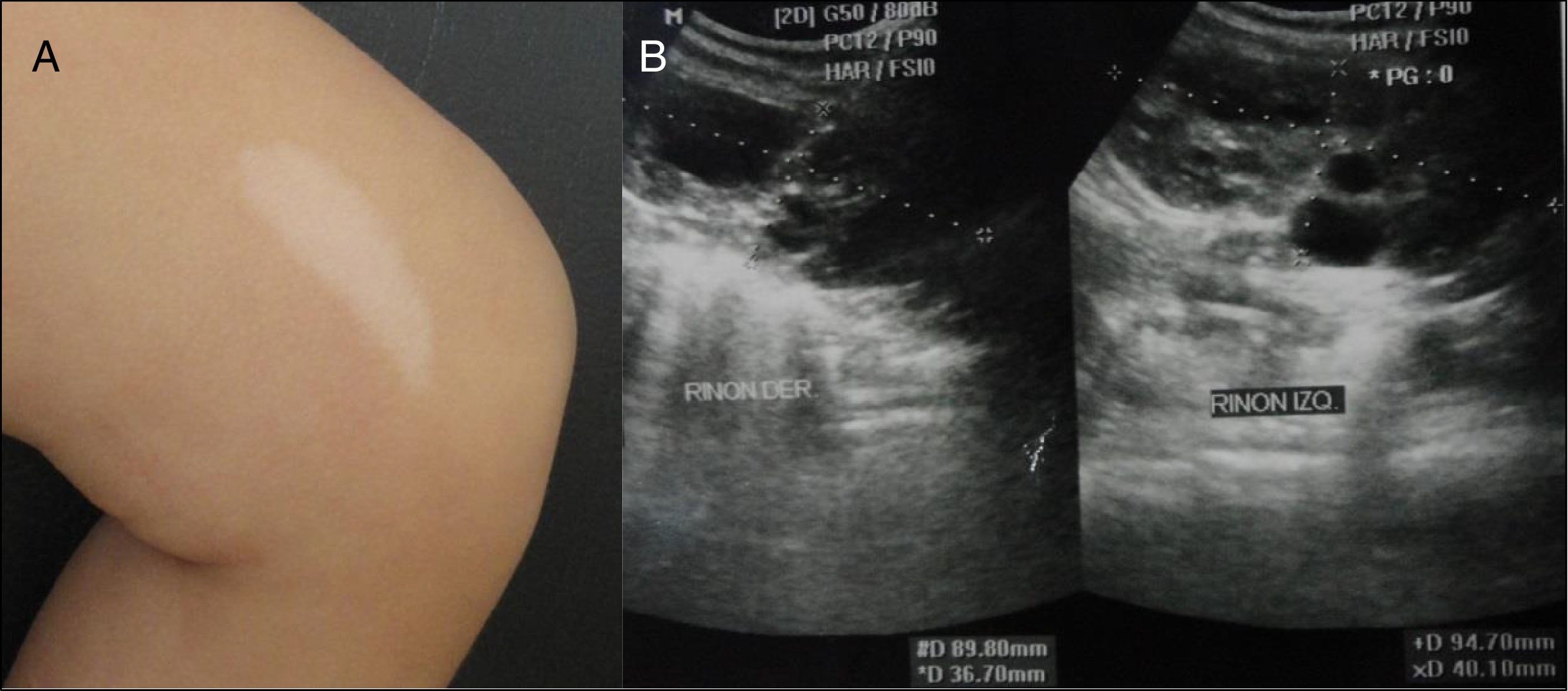
Al examen físico presenta tendencia a la dolicocefalia con facies peculiar caracterizada por prominencia frontal, el puente nasal es cóncavo con punta aplanada y narinas antevertidas, el filtrum es corto y ancho. Presenta 7 lesiones hipocrómicas, 2 en hoja de fresno que corresponde las más grandes ubicadas en región lumbar y muslo izquierdo (fig. 1a).
 Lesión hipocrómica en forma de hoja de fresno en muslo izquierdo; b) La ecografía renal muestra aumento de volumen en ambos riñones y múltiples quistes bilaterales.")
La resonancia magnética nuclear evidencia áreas multifocales con comportamiento hiperintenso en T2 flair, en forma de parches, a nivel de ambos hemisferios parietales que no condicionan efecto de masa sobre estructuras circunscritas. El electroencefalograma en sueño fue anormal por la presencia de actividad paroxística, brotes de puntas y ondas agudas en áreas fronto polar y temporal derecha. La ecografía ocular mostró lesión elevada con estructura interna hipoecogénica en área papilar, compatible con hamartoma astrocítico en nervio óptico de ojo derecho. La ecocardiografía transtorácica evidencia tumoración en tracto de salida en ventrículo derecho sugestivo de rabdomioma que no ocasiona obstrucción, ni repercusión hemodinámica. La ecografía renal aumentó de volumen en ambos riñones, 9,4×4,0cm para el riñón izquierdo y 9×3,6cm para el derecho. Presenta formación quística de contenido líquido, algunos con paredes gruesas, el de mayor tamaño en el polo inferior en el riñón derecho de 6×3,1cm (fig. 1b). El perfil renal se encuentra dentro de los valores de referencia, así como otros estudios séricos.
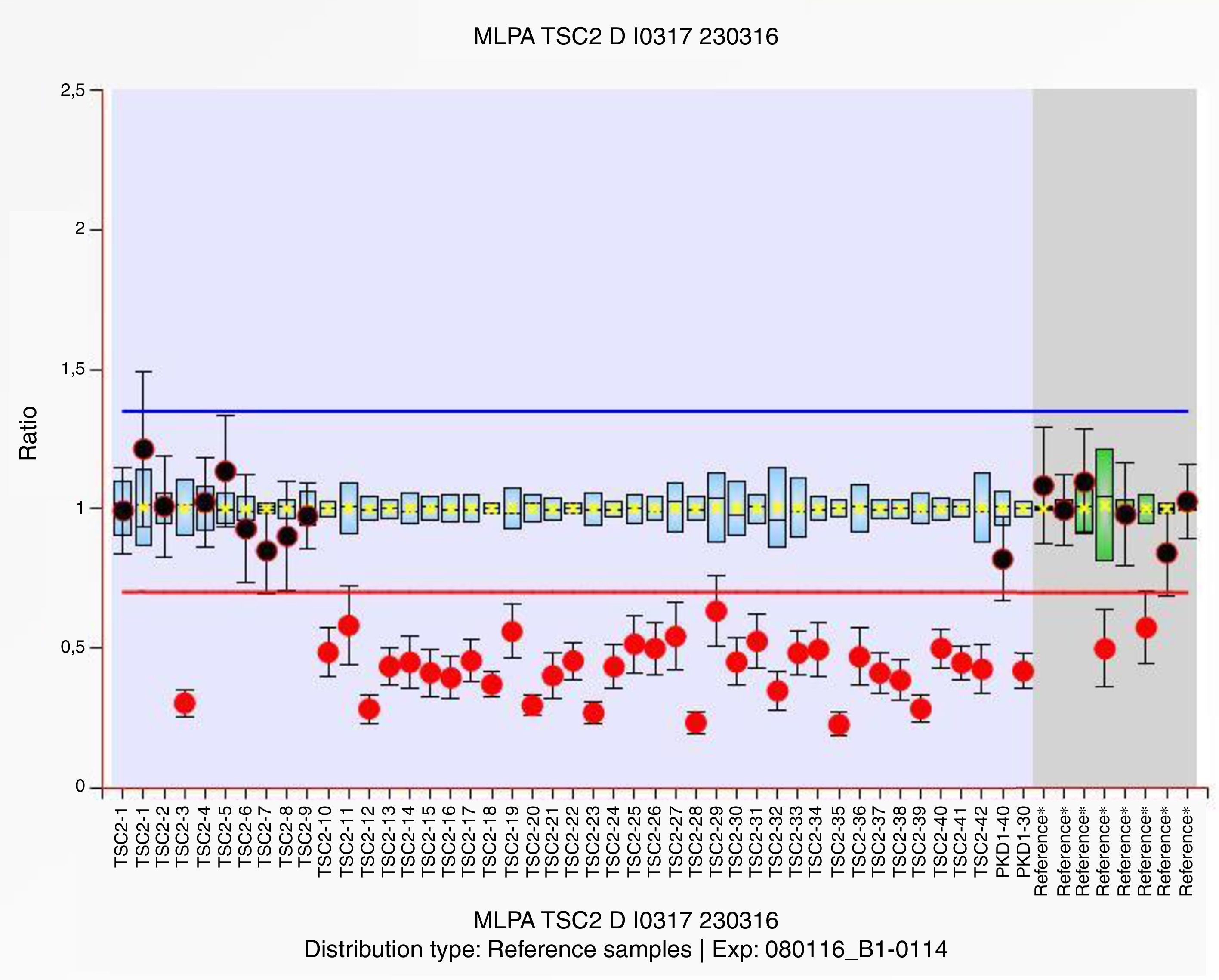
Se extrajo ADN y en la secuenciación masiva en la plataforma MiSeq (Illumina) de los genes TSC1 y TSC2 se encontraron 2 polimorfismos en heterocigosis en TSC1, GAA/GAG, c.1335A>G, p.Glu445Glu y ATG/ACG, c.965T>C, p.Met322Thr con referencias rs7862221 y rs1073123, respectivamente. El estudio de amplificación de sondas dependiente de ligandos múltiples (MLPA P337) mostró una deleción en heterocigosis de las sondas localizadas en los exones 10 al 42 del gen TSC2, así como en los exones 30 y 40 de del gen PKD1 (fig. 2).

El complejo esclerosis tuberosa (OMIM #191100 y #613254) es un trastorno multisistémico que exhibe un patrón de herencia autosómico dominante, caracterizado por la presencia de múltiples hamartomas a nivel de cerebro, ojos, corazón, riñones y piel1–3. Se caracteriza por un amplio espectro fenotípico y variable que incluye convulsiones, retardo mental, alteraciones renales y en piel, además de un aumento del riesgo de malignidad2,4. Su incidencia es de uno entre 6.000 y 11.000 nacidos vivos1. Se debe a mutaciones en los genes supresores de tumores TSC1 (OMIM #605284) y TSC2 (OMIM #191092), que codifican a la hamartina y la tuberina, respectivamente1,3. Este segundo gen se encuentra localizado en 16p13.3 adyacente al gen PKD1 (OMIM #601313), responsable del 85% de los casos de poliquistosis renal (OMIM #173900), que igualmente presentan un patrón de herencia autosómico dominante1,5. Este es el trastorno renal hereditario más frecuente, con una incidencia de uno entre 400 y 1.000 nacidos vivos5.
Una deleción de gran tamaño que puede abarcar los genes TSC2 y PKD1 produce el llamado síndrome de genes contiguos TSC2/PKD1 (OMIM #600273)1,2,5. Fue descrito por primera vez por Brook-Carter et al. en 1994, en 6 pacientes con complejo esclerosis tuberosa con poliquistosis renal severa infantil6. Se caracteriza por la presencia de quistes renales grandes, bilaterales, congénitos o de aparición muy precoz, lo que puede modificar su pronóstico. Se estima que el 5% de los pacientes con complejo de esclerosis tuberosa presentan poliquistosis renal1.
Las complicaciones renales suponen la segunda causa de muerte posterior a la afectación neurológica en el complejo de esclerosis tuberosa. Los angiomiolipomas son la alteración renal más frecuente en adultos y en niños, y pueden estar presentes en 16% de los casos, además de los quistes de pequeño tamaño y el carcinoma de células renales1,3.
Presentamos el caso de una niña con diagnóstico genético y clínico de síndrome de genes contiguos TSC2/PKD1 resaltando la valoración multidisciplinaria ante el pleitropismo y la gravedad de la entidad, así como impartir oportuno asesoramiento genético con un riesgo de recurrencia del 50% en los descendientes de los individuos afectados.



