














El síndrome hemolítico urémico (SHU) es una entidad clínica definida por la tríada anemia hemolítica no inmune, trombocitopenia e insuficiencia renal aguda, en la que las lesiones subyacentes están mediadas por un proceso de microangiopatía trombótica (MAT) sistémico. Distintas causas pueden desencadenar el proceso de MAT que caracteriza el SHU. En este documento consideramos SHU atípico (SHUa) como el subtipo de SHU en el que los fenómenos de MAT son fundamentalmente consecuencia del daño producido en el endotelio de la microvasculatura renal y de otros órganos por desregulación de la actividad del sistema del complemento. En los últimos años se han identificado diversas mutaciones en genes del sistema del complemento asociados a SHUa, que explicarían aproximadamente el 60% de los casos de SHUa, y se han caracterizado funcionalmente numerosas mutaciones y polimorfismos asociados a SHUa que han permitido determinar que la patología se produce como consecuencia de la deficiente regulación de la activación del complemento sobre las superficies celulares y que lleva al daño endotelial mediado por la activación del C5 y de la vía terminal del complemento. Eculizumab es un anticuerpo monoclonal humanizado que inhibe la activación del C5, bloqueando la generación de la molécula proinflamatoria C5a y la formación del complejo de ataque de membrana. En estudios prospectivos en pacientes con SHUa su administración ha demostrado la interrupción rápida y sostenida del proceso de MAT, con una mejora significativa de la función renal a largo plazo y una reducción importante de la necesidad de diálisis y el cese de la terapia plasmática. En función de las evidencias científicas publicadas y la experiencia clínica acumulada, el Grupo Español de SHUa publicamos un documento de consenso con recomendaciones para el tratamiento de la enfermedad (Nefrología 2013;33(1):27-45). En la presente versión online del documento se actualizan los contenidos sobre la clasificación etiológica de las MAT, la fisiopatología del SHUa, su diagnóstico diferencial y su manejo terapéutico.
Haemolytic uraemic syndrome (HUS) is a clinical entity defined as the triad of nonimmune haemolytic anaemia, thrombocytopenia, and acute renal failure, in which the underlying lesions are mediated by systemic thrombotic microangiopathy (TMA). Different causes can induce the TMA process that characterizes HUS. In this document we consider atypical HUS (aHUS) a sub-type of HUS in which the TMA phenomena are the consequence of the endotelial damage in the microvasculature of the kidneys and other organs due to a disregulation of the activity of the complement system. In recent years, a variety of aHUs-related mutations have been identified in genes of the the complement system, which can explain approximately 60% of the aHUS cases, and a number of mutations and polymorphisms have been functionally characterized. These findings have stablished that aHUS is a consequence of the insufficient regulation of the activiation of the complement on cell surfaces, leading to endotelial damage mediated by C5 and the complement terminal pathway. Eculizumab is a monoclonal antibody that inhibits the activation of C5 and blocks the generation of the pro-inflammatory molecule C5a and the formation of the cell membrane attack complex. In prospective studies in patients with aHUS, the use of Eculizumab has shown a fast and sustained interruption of the TMA process and it has been associated with significative long-term improvements in renal function, the interruption of plasma therapy and important reductions in the need of dialysis. According to the existing literature and the accumulated clinical experience, the Spanish aHUS Group published a consensus document with recommendations for the treatment of aHUs (Nefrologia 2013;33[1]:27-45). In the current online version of this document, we update the aetiological classification of TMAs, the pathophysiology of aHUS, its differential diagnosis and its therapeutic management.
El síndrome hemolítico urémico (SHU) es una entidad clínica que se define por la tríada anemia hemolítica microangiopática no inmune, trombocitopenia e insuficiencia renal aguda1. Las lesiones histológicas del SHU se caracterizan por la aparición de microangiopatía trombótica (MAT) sistémica, que afecta preferentemente a los vasos intrarrenales. La mayoría de los casos de SHU son causados por una infección entérica por Escherichia coli productora de toxina Shiga (STEC) u otros gérmenes productores de verotoxina (VTEC), dando lugar a lo que se conoce como SHU típico o STEC (VTEC)-SHU. En aproximadamente un 10% de los casos el SHU se produce como consecuencia de una desregulación de la vía alternativa del sistema del complemento, por causas genéticas o adquiridas (autoanticuerpos), que conduce al desarrollo de daño endotelial y fenómenos de MAT sistémica2. Este tipo de SHU relacionado con una desregulación del complemento se denomina SHU atípico (SHUa).
En el año 2011 las agencias reguladoras de Estados Unidos y Europa aprobaron la indicación de Eculizumab (Soliris®; Alexion Pharmaceuticals, Connecticut, EE.UU.) para el tratamiento del SHUa3. Eculizumab es un anticuerpo monoclonal humanizado que actúa inhibiendo la activación del C5 y bloqueando la generación de la anafilatoxina proinflamatoria C5a y la formación del complejo de ataque de membrana, causante de la lisis celular4. En estudios prospectivos en pacientes con SHUa, Eculizumab ha demostrado interrumpir eficazmente el proceso de MAT y sus consecuencias, asociándose con una rápida y significativa mejora de las alteraciones hematológicas y de la función renal, que se mantiene a largo plazo5, así como con mejoras de la afectación sistémica y de la hipertensión arterial.
En 2012, el Grupo Español de SHUa celebramos una reunión de consenso para la elaboración de un documento con recomendaciones para el tratamiento de la enfermedad6. Desde entonces, el grupo ha venido realizando reuniones anuales con el objetivo de actualizar, en base a las evidencias científicas publicadas y la experiencia clínica, el conocimiento sobre los diversos aspectos de interés de la enfermedad (tales como la clasificación etiológica de las MAT, la fisiopatología del SHUa y el diagnóstico diferencial), así como las recomendaciones de tratamiento. En la presente versión online del documento de consenso se actualizan los contenidos publicados originalmente en Nefrología 2013;33(1):27-45.
Clasificación etiológica de las microangiopatías trombóticasEl término MAT define una lesión histológica de arteriolas y capilares que se caracteriza por engrosamiento e inflamación de la pared vascular, desprendimiento de células endoteliales, ampliación del espacio subendotelial por acumulaciones de proteínas y material de lisis celular, y la presencia de trombos plaquetarios ocluyendo las luces vasculares1 (fig. 1). Existen 2 entidades clínicas caracterizadas por lesiones de MAT primaria, de causa y base fisiopatológica diferente: la púrpura trombótica trombocitopénica (PTT) y el SHU.
Glomérulos isquémicos y retraídos. B)Mesangiólisis. C)Trombos en los capilares glomerulares (flecha). D)Arteriola ocluida por trombos plaquetarios. Fotos cortesía de la Dra. R. Ortega (Servicio de Anatomía Patológica, Hospital Universitario Reina Sofía, Córdoba).")
Lesiones histopatológicas renales del síndrome hemolítico urémico. A)Glomérulos isquémicos y retraídos. B)Mesangiólisis. C)Trombos en los capilares glomerulares (flecha). D)Arteriola ocluida por trombos plaquetarios.
Fotos cortesía de la Dra. R. Ortega (Servicio de Anatomía Patológica, Hospital Universitario Reina Sofía, Córdoba).
La trombosis intravascular en la PTT es consecuencia de una deficiencia grave de la actividad metaloproteasa de A Desintegrin and Metalloproteninasa with ThromboSpondin type 1 motif, member 13 (ADAMTS13), una enzima plasmática encargada de fragmentar los multímeros ultralargos del factor de Von Willebrand7. Dicha deficiencia puede ser de causa genética o adquirida por anticuerpos circulantes de tipo IgG que bloquean ADAMTS13 (especialmente en pacientes en tratamiento con antiagregantes plaquetarios)8.
El 90% de los casos de SHU son causados por una infección entérica por STEC a partir de alimentos contaminados (SHU típico o STEC [VTEC]-SHU)2. La toxina Shiga ejerce un efecto lesivo directo sobre el endotelio vascular, desencadenando diversos eventos celulares y vasculares que conducen al desarrollo de MAT2. Clínicamente suele comenzar con dolor abdominal y diarrea, desarrollándose a los 4-10días un fracaso renal agudo. El pronóstico suele ser favorable, siendo la mortalidad inferior al 5% y observándose la recuperación clínica completa en el 80% de los pacientes, si bien hasta el 20-30% de los pacientes progresan a insuficiencia renal crónica grave a largo plazo9,10.
El diagnóstico de SHUa es esencialmente por exclusión, una vez se descarte un déficit de ADAMTS13 (PTT) o la infección por STEC (STEC-SHU). En los pacientes con SHUa los fenómenos de MAT son consecuencia de la desregulación de la vía alternativa del complemento sobre las superficies celulares. Esta alteración, en la que se identifican factores genéticos o autoanticuerpos en un porcentaje creciente de casos, impide que cuando el complemento se activa (por diversos factores desencadenantes) se controle adecuadamente la actividad sobre células propias, provocando daño endotelial, inflamación y trombosis secundaria. De los más de 1.000 pacientes con SHUa publicados en la literatura se han detectado mutaciones en una o más proteínas del sistema del complemento en aproximadamente el 60% de ellos11-18, aunque no se descarta que en el resto exista también un componente genético (con implicación de genes del complemento u otros tipos, como genes de la coagulación) y/o de autoinmunidad no identificado. De hecho, es destacable el hallazgo de autoanticuerpos contra el factor H del complemento (FH) en el 5-10% de los pacientes con SHUa19. A diferencia del STEC-SHU, que suele ser un evento único, el SHUa es una entidad crónica y recidivante desencadenada por activaciones incontroladas del sistema del complemento. Antes de la disponibilidad de Eculizumab, el SHUa se asociaba en la mayoría de los casos con mal pronóstico: la mortalidad tras un primer episodio de SHUa era del 10-15%, y hasta un 50% de los pacientes no recuperaban la función renal11,12,20.
Recientemente se ha descrito un tipo de SHUa producido por mutaciones recesivas en el gen DGKE que codifica la proteína DGK-¿ (diacilglicerol quinasa-¿)21. La pérdida de actividad de esta enzima, presente en células endoteliales, plaquetas y podocitos, induce apoptosis de las células endoteliales y deteriora la respuesta angiogénica, lo que lleva a un estado protrombótico e inflamatorio22. Los pacientes con mutaciones DGKE exhiben diferentes fenotipos que van desde el SHUa a una glomerulonefritis membranoproliferativa con elevada proteinuria y síndrome nefrótico23. Los individuos afectos de SHUa presentan en el primer año de vida hipertensión arterial persistente y hematuria-proteinuria (incluso en rango nefrótico). En estos casos, y en contraste con lo que se observa en los casos pediátricos de SHUa asociados con alteraciones genéticas del complemento, la evolución a la enfermedad renal crónica no es abrupta, sino progresiva en años21.
Por otra parte, además de la infección entérica por STEC (SHU típico), de las alteraciones en la regulación de la activación del complemento, de las mutaciones en DGKE o genes de la coagulación (SHUa) o del déficit de ADAMTS13 (genético o autoinmune) en la PTT, existen otros factores y entidades clínicas que pueden asociarse con el desarrollo de MAT. Englobamos este tipo de MAT bajo el término de MAT secundarias. En niños, algunos casos se asocian con aciduria metilmalónica24 o, más frecuentemente (5% de los casos de SHU en niños), con infecciones invasivas por serotipos de Streptococcus pneumoniae productores de la enzima neurominidasa (que exponen el criptoantígenoT en la superficie celular y originan el fenómeno de MAT25), o con la infección por el virus H1N126. En general, se han descrito casos de MAT asociados a infecciones víricas (CMV, VIH, parvovirus), procesos neoplásicos, fármacos (antitumorales como los inhibidores del factor de crecimiento vascular endotelial, inmunosupresores como los inhibidores de la calcineurina [ciclosporina y tacrolimus] o los inhibidores de la mammalian target of rapamycin [mTOR; sirolimus, everolimus], antiagregantes plaquetarios, antivirales u anticonceptivos orales, entre otros), la hipertensión arterial maligna, el trasplante de médula ósea o de órganos sólidos, el embarazo y el posparto, enfermedades sistémicas autoinmunes o glomerulonefritis27.
Es importante recalcar que en algunos pacientes no es posible hallar ninguna de las mencionadas causas de MAT, mientras que en otros puede coexistir más de un factor etiológico, dando lugar a una clínica heterogénea y a un diagnóstico difícil. De hecho, el solapamiento entre estas entidades no es infrecuente y se ha descrito, por ejemplo, que hasta un 25% de los pacientes con STEC-SHU y un 86% de las pacientes con SHU asociado a embarazo presentan mutaciones en el sistema del complemento, pudiéndose considerar en estos casos que la enfermedad subyacente es en realidad un SHUa28,29. También se han encontrado mutaciones en el sistema del complemento en un 27% de los pacientes con SHU postrasplante asociado al uso de inhibidores de la calcineurina y en un 33% de los pacientes con SHU asociado a enfermedades autoinmunes12. Adicionalmente, hasta la fecha se han reportado varios casos de pacientes con MAT secundarias tratados de forma exitosa con Eculizumab (MAT asociada a fármacos30, a trasplante de órgano sólido31 o de médula ósea32, a embarazo33 y a lupus eritematoso sistémico34). El hecho de que el bloqueo del complemento (Eculizumab) se asocie con una buena respuesta clínica y una reversibilidad de la MAT sugiere que la desregulación del complemento de base no genética tiene probablemente un papel importante en muchos casos de MAT secundarias, predisponiendo a los pacientes a su desarrollo. En función de todo lo anterior, la figura 2 presenta una propuesta de clasificación etiológica de las MAT y una representación del potencial solapamiento que puede tener lugar entre estas entidades clínicas. Resulta necesario subrayar que la clasificación de las MAT es un tema de actualidad y que existe un importante debate en la comunidad médica como consecuencia del constante avance en el conocimiento de la fisiopatología de estas entidades35. Como el motivo de discusión principal de este documento es la actualización del SHUa mediado por desregulación del complemento, en los siguientes apartados únicamente se hace referencia a esta entidad.

Clasificación etiológica de las microangiopatías trombóticas.
ADAMTS13: A Disintegrin And Metalloproteinase with a ThromboSpondin type 1 motif, member 13; CMV: citomegalovirus; FB: factor B del complemento; FH: factor H del complemento; FI: factor I del complemento; HELLP: Hemolysis, Elevated Liver enzymes, Low Platelet count; LES: lupus eritematoso sistémico; MAT: microangiopatía trombótica; MCP: proteína cofactor de membrana; mTOR: mammalian target of Rapamycin; PTT: púrpura trombótica trombocitopénica; SHU: síndrome hemolítico urémico; SHUa: síndrome hemolítico urémico atípico; STEC: Escherichia coli productor de toxina Shiga; THBD: trombomodulina; VEGF: factor de crecimiento vascular endothelial; VHC: virus de la hepatitis C; VIH: virus de la inmunodeficiencia humana.
El SHUa se considera una enfermedad ultrarrara. Existen muy pocos datos acerca de su incidencia y prevalencia, siendo limitados el conocimiento de la epidemiología real de enfermedad. En Estados Unidos se estima que el SHUa tiene una incidencia anual de ∼1-2casos/millón de habitantes36. En Europa, en un estudio multicéntrico internacional reciente se observó una incidencia de 0,11 casos/millón de habitantes. Con relación a la prevalencia, la European Medicines Agency (EMA) estima que esta puede ser ∼3,3pacientes por millón de habitantes/año en menores de 18años, con cifras inferiores en adultos.
El SHUa afecta mayoritariamente a niños y adultos jóvenes, aunque puede aparecer en cualquier edad de la vida11,12. El inicio de la enfermedad es más frecuente antes de los 18años (60% vs. 40%), siendo la distribución por sexos similar (con cierta preponderancia en mujeres cuando la enfermedad aparece en la edad adulta)11,13.
ClínicaEl inicio de la clínica suele ser abrupto, aunque en un 20% de los pacientes puede ser progresivo (semanas o meses) con anemia subclínica, trombocitopenia fluctuante y función renal conservada11. El cuadro se caracteriza por la tríada de anemia hemolítica microangiopática no inmune, trombocitopenia y fracaso renal agudo1. Los niveles altos de lactato deshidrogenasa (LDH), los niveles indetectables de haptoglobina y la observación de esquistocitos confirman la presencia de hemólisis intravascular20 asociada a hematuria, proteinuria y/o fracaso renal agudo (con o sin oligoanuria). La incidencia de hipertensión arterial, por sobrecarga de volumen o por lesión vascular, es frecuente1. En algunos pacientes la única manifestación de MAT puede ser proteinuria con hipertensión arterial y desarrollo de insuficiencia renal progresiva sin alteraciones hematológicas.
Aunque las lesiones en el SHUa afectan predominantemente a los vasos renales, el carácter difuso y sistémico del fenómeno de MAT conduce a la afectación de la microvasculatura de otros órganos (cerebro, corazón, intestino, páncreas y pulmones, entre otros)1, lo que explica la aparición frecuente de síntomas extrarrenales11,12. Los más frecuentes son los de tipo neurológico (48%)37, incluyendo irritabilidad, somnolencia, confusión, convulsiones, encefalopatía, accidente cerebrovascular, hemiparesias, alteraciones visuales, hemiplejías o coma1,12,37,38. El infarto de miocardio se ha descrito hasta en un 3% de los pacientes con SHUa, pudiéndose relacionar con muerte súbita12,39. La miocardiopatía, la insuficiencia cardiaca y la vasculopatía isquémica periférica también han sido descritas19,37,40,41, así como la diarrea (30%) y otros síntomas gastrointestinales (colitis, náuseas, vómitos, dolor abdominal, hepatitis, colestasis y pancreatitis, entre otros)12,19,38,42. Recientemente se han reportado casos de pacientes con SHUa con afectación cutánea en forma de lesiones ulcerosas en las extremidades inferiores43. La variabilidad de la sintomatología dificulta el diagnóstico diferencial con otras causas de MAT.
FisiopatologíaEl sistema del complemento, formado por numerosas proteínas plasmáticas circulantes y asociadas a las membranas celulares, forma parte de la inmunidad innata y es esencial en la defensa contra las infecciones, el procesamiento de complejos inmunes, la respuesta de anticuerpos y la eliminación de restos apoptóticos. Su activación por cualquiera de las vías existentes (clásica, lectina y alternativa) conlleva la formación de complejos multiproteicos con actividad C3-convertasa que escinden la proteína C3, generando C3b (fig. 3). Esta molécula puede unirse covalentemente a las superficies responsables de la activación del complemento, facilitando su fagocitosis por leucocitos polimorfonucleares y macrófagos e iniciando la activación de C5 que lleva al ensamblaje del complejo de ataque a la membrana que conduce a la lisis celular. Además, como el C3b es uno de los componentes de la C3-convertasa de la vía alternativa, su generación amplifica exponencialmente la activación del complemento promoviendo la formación de más C3-convertasas44. Para evitar que la activación del complemento lo consuma totalmente e impedir dañar los tejidos propios (el C3b se une indiscriminadamente tanto a patógenos como a células propias), existen numerosas proteínas reguladoras del proceso, como el FH, la proteína cofactor de membrana (MCP) y el factori del complemento (FI), que disocian las C3-convertasas e inducen la degradación de C3b. En consecuencia, en condiciones normales los niveles de C3b se mantienen bajos y cuando se activa el complemento su depósito se limita a las estructuras responsables de esa activación.
 lleva a que se depositen grandes cantidades de C3b sobre la membrana celular del activador, lo que conduce a su opsonización y a la activación del C5 (vía terminal o lítica), que conduce a la formación del complejo de ataque a la membrana y la lisis celular. La activación del complemento produce inflamación y reclutamiento de leucocitos. El proceso central en la activación del complemento es la generación de C3b. Su formación depende de complejos enzimáticos inestables llamados C3-convertasas, que catalizan la rotura de C3 para generar C3b. C3b, a su vez, es capaz de formar más C3-convertasa de la vía alternativa (C3bBb), amplificando así la activación inicial. La generación de C3b está regulada a 2 niveles: disociación de las C3-convertasas e inactivación proteolítica del C3b y C4b. Varias proteínas reguladoras en plasma y en la membrana celular llevan a cabo esta regulación. Entre ellas, factor H, MCP y factor I desempeñan un papel fundamental en la disociación de la C3-convertasa de la vía alternativa (C3bBb) y en la degradación proteolítica de C3b. Las mutaciones en estas proteínas encontradas en pacientes con SHUa interfieren esta función reguladora de la activación de la vía alternativa. Algunos pacientes con SHUa son portadores de mutaciones en las proteínas C3 y factor B que organizan la C3-convertasa. Estas mutaciones son particulares en el sentido de que aumentan la actividad de las proteínas mutadas (son mutaciones ganancia de función), lo que resulta en un aumento en la activación del complemento que excede la capacidad de las proteínas reguladoras.")
Desregulación del complemento en el síndrome hemolítico urémico atípico. La activación del complemento por cualquiera de las 3 vías (reconocimiento de antígenos extraños, vía alternativa; de anticuerpos, vía clásica; o de polisacáridos de manano, vía de las lectinas) lleva a que se depositen grandes cantidades de C3b sobre la membrana celular del activador, lo que conduce a su opsonización y a la activación del C5 (vía terminal o lítica), que conduce a la formación del complejo de ataque a la membrana y la lisis celular. La activación del complemento produce inflamación y reclutamiento de leucocitos. El proceso central en la activación del complemento es la generación de C3b. Su formación depende de complejos enzimáticos inestables llamados C3-convertasas, que catalizan la rotura de C3 para generar C3b. C3b, a su vez, es capaz de formar más C3-convertasa de la vía alternativa (C3bBb), amplificando así la activación inicial. La generación de C3b está regulada a 2 niveles: disociación de las C3-convertasas e inactivación proteolítica del C3b y C4b. Varias proteínas reguladoras en plasma y en la membrana celular llevan a cabo esta regulación. Entre ellas, factor H, MCP y factor I desempeñan un papel fundamental en la disociación de la C3-convertasa de la vía alternativa (C3bBb) y en la degradación proteolítica de C3b. Las mutaciones en estas proteínas encontradas en pacientes con SHUa interfieren esta función reguladora de la activación de la vía alternativa. Algunos pacientes con SHUa son portadores de mutaciones en las proteínas C3 y factor B que organizan la C3-convertasa. Estas mutaciones son particulares en el sentido de que aumentan la actividad de las proteínas mutadas (son mutaciones ganancia de función), lo que resulta en un aumento en la activación del complemento que excede la capacidad de las proteínas reguladoras.
Numerosos estudios han establecido que aproximadamente un 60% de los pacientes con SHUa son portadores de mutaciones en los genes reguladores del complemento (CFH, MCP, CFI, trombomodulina [THBD], o en los componentes de la C3-convertasa, factor B [FB] y C3)45-54. Todas estas mutaciones causan desregulación de la vía alternativa (tabla 1). El FH actúa en plasma controlando la homeostasis del complemento y sobre superficies celulares evitando el daño a componentes propios. Las mutaciones en la región C-terminal de FH son características de SHUa. Estas mutaciones, al alterar una región de FH que es necesaria para regular la activación del complemento sobre las superficies celulares, disminuyen la protección de las células al daño accidental producido por la activación del complemento, pero no afectan la regulación del complemento en plasma55. Del mismo modo, el ensayo funcional de las mutaciones asociadas con SHUa encontradas en otros genes del complemento, como MCP, CFI, CFB o C3, ha confirmado que todas ellas causan un defecto en la protección de las superficies celulares y que esta pérdida de regulación del complemento puede deberse a una disminución en la actividad de las proteínas reguladoras o a una actividad anormalmente elevada de las C3-convertasas. Así, mientras que las mutaciones en FH, MCP y FI incapacitan a estas proteínas para realizar su función reguladora, las mutaciones en FB o C3 resultan en una C3-convertasa más activa.
Factores de riesgo en el síndrome hemolítico urémico atípicoa
| Mutaciones |
| Pérdida de función |
| CFH (∼ 13%) |
| MCP (∼ 11%) |
| CFI (∼ 10%) |
| THBD (∼ 4%) |
| Ganancia de función |
| C3 (∼ 4%) |
| CFB (∼ 3%) |
| Polimorfismos |
| Aumentan riesgo |
| CFH: c.-332C>T; c.2016A>G (p.Gln672Gln); c.2808G>T (p.Glu936Asp) |
| MCP: c.-652A>G; c.-366A>G; c.989-78G>A; *897T>C |
| Confieren protección |
| CFH: c.184G>A (p.Val62Ile) |
| Autoanticuerpos |
| Anti-FH (∼ 5%) |
| Factores ambientales |
| Infecciones |
| Fármacos inmunosupresores |
| Anticonceptivos orales |
| Fármacos anticancerosos |
Anti-FH: anticuerpos anti-factor H del complemento; CFB: gen del factor B del complemento; CFH: gen del factor H del complemento; CFI: gen del factor i del complemento; MCP: gen de la proteína cofactor de membrana; THBD: gen de la trombomodulina.
Teoría de los «multiple hits». El SHUa es una enfermedad compleja en la que normalmente se combinan diferentes factores de riesgo, genéticos y ambientales. No es raro que los pacientes sean portadores de más de una mutación en genes del complemento, o que combinen mutaciones con polimorfismos de riesgo. Además, son necesarios también factores ambientales que contribuyan a poner de manifiesto la predisposición genética que aportan las mutaciones o los polimorfismos. La concurrencia de una mutación con otras mutaciones, con polimorfismos de riesgo, con autoanticuerpos o con factores ambientales desencadenantes, explica la penetrancia incompleta del SHUa, así como las diferencias en su presentación y evolución, entre portadores de mutaciones en genes del complemento.
Un 5-10% de los pacientes con SHUa presentan autoanticuerpos anti-FH dirigidos contra la región C-terminal, con consecuencias similares a las de las mutaciones en FH56,57. Su papel en la patogénesis del SHUa no está completamente establecido, pero parecen asociarse con el inicio o las recurrencias de la enfermedad. El título de anticuerpos puede disminuir con el tiempo, debiéndose realizar su búsqueda al inicio del SHUa. La presencia de anticuerpos anti-FH se asocia en los pacientes con SHUa con el déficit completo de la proteína1 relacionada con el FH (FHR1)58.
La penetrancia del SHUa en los portadores de mutaciones en alguno de los genes del complemento es del 50% aproximadamente, siendo habitual que en familias con mutaciones identificadas solo algunos de los portadores desarrollen SHUa y que incluso la presentación clínica en estos individuos sea distinta. Existe también una gran heterogeneidad clínica entre pacientes no emparentados portadores de la misma mutación. Esto es debido a la existencia de factores de riesgo adicionales (genéticos y ambientales) que modulan el desarrollo y la evolución de la enfermedad. La búsqueda de mutaciones del complemento en pacientes con SHUa y la realización de estudios de asociación casos-controles utilizando polimorfismos genéticos en genes candidatos o marcadores genéticos distribuidos a lo largo del genoma humano ha identificado que algunas variantes (polimorfismos) de los genes CFH y MCP modulan la penetrancia y la gravedad de la enfermedad (tabla 1)49,59,60.
Los haplotipos CFH-H3 y MCPggaac son los polimorfismos más relevantes asociados con riesgo de SHUa. Ambos haplotipos incluyen polimorfismos de un solo nucleótido (single nucleotyde polymorphysms [SNP]) localizados en la región promotora de los genes CFH y MCP que disminuyen la expresión de FH y MCP. La presencia de ambos polimorfismos en homocigosis podría justificar la predisposición a SHUa en individuos en los que no se han encontrado mutaciones en ninguno de los genes asociados con SHUa. Un estudio colaborativo reciente realizado por el European Working Party on Complement Genetics in Renal Diseases en 795 pacientes con SHUa ha identificado que el 3% de estos pacientes son portadores de mutaciones combinadas en más de un gen. Además, este extenso estudio ha puesto de manifiesto que la presencia concomitante de los haplotipos de riesgo CFH-H3 y MCPggaac también aumenta significativamente la penetrancia de la enfermedad en portadores de mutaciones combinadas, reforzando la conclusión de que el genotipado de estos polimorfismos de riesgo ayuda a predecir el riesgo de desarrollar SHUa en portadores de mutaciones61.
De forma adicional a las mencionadas alteraciones genéticas, en el inicio del SHUa participan también factores ambientales desencadenantes. Las mutaciones comentadas anteriormente predisponen a la enfermedad impidiendo una regulación adecuada del complemento sobre las superficies celulares ante una situación que dispare la activación del sistema en la microvasculatura. Los eventos infecciosos desencadenan el SHUa en el 50-80% de los pacientes11,12,40, especialmente los del tracto respiratorio superior (virus de la influenza H1N1). La diarrea por gastroenteritis puede preceder al SHUa hasta un 30% de los casos12 (incluyendo diarrea por STEC11,12,19). En mujeres, el embarazo, en particular el periodo posparto, es un frecuente factor desencadenante de SHUa12,29, así como el uso de anovulatorios orales.
Mutaciones en el gen que codifica trombomodulina (THBD), una proteína anticoagulante que actúa como cofactor de la trombina y que también regula la inactivación de C3b mediada por FI, se han asociado con SHUa62. Acorde con la desregulación del complemento que caracteriza a los pacientes con SHUa, el análisis funcional de las mutaciones en THBD asociadas con SHUa ha demostrado que las mutaciones en trombomodulina alteran su actividad reguladora del complemento62. Sin embargo, se desconoce si las mutaciones en trombomodulina asociadas con SHUa alteran también la actividad anticoagulante y, por lo tanto, si alteraciones de esta actividad pudieran ser también relevantes en SHUa. En este sentido, un estudio reciente en 36 pacientes con SHUa ha evaluado mediante secuenciación de ADN masiva la presencia de mutaciones en los genes del sistema del complemento y de la coagulación, encontrando mutaciones en genes de ambos sistemas63. El gen del sistema de la coagulación que presentaba mayor número de mutaciones fue el del plasminógeno (PLG), un cimógeno que, convertido en plasmina, juega un importante papel en la fibrinólisis. Aunque estos datos sugieren una contribución de los genes de la coagulación en la predisposición a SHUa (en particular de PLG), son necesarios estudios adicionales que confirmen estas observaciones.
La búsqueda de nuevos genes asociados con SHUa ha sido también abordada por Lemaire et al.21 utilizando la secuenciación de exomas. Estos autores han identificado mutaciones en homocigosis en el gen DGKE que codifica la proteína DGK-¿ en 13 pacientes con SHUa, pertenecientes a 9 familias. Estos pacientes tuvieron un comienzo muy temprano del SHUa, normalmente durante el primer año de vida, seguido de múltiples recurrencias y evolución frecuente a fracaso renal terminal en la segunda década de la vida21. Recientemente se ha establecido que el déficit de DGK-¿ en células endoteliales induce la expresión de ICAM-1 y factor tisular a través de un aumento de la señalización mediada por p36-MAPK, lo que induce apoptosis, altera la respuesta angiogénica y determina un fenotipo proinflamatorio y protrombótico. Sin embargo, la ausencia de DGK-¿ no afecta la activación del complemento en las superficies celulares21,22. Es muy probable que la ausencia de DGK-¿ en podocitos y células endoteliales altere el diafragma de filtración glomerular, lo que explicaría la presencia de proteinuria masiva y la susceptibilidad que manifiestan estos pacientes a desarrollar patología glomerular21,23, aunque se desconoce por qué en estos pacientes se desarrollen con frecuencia diferentes patologías glomerulares. Finalmente, aunque el papel del complemento en el desarrollo de la enfermedad renal en los portadores de mutaciones en DGKE se había descartado inicialmente21, Hace poco se han identificado pacientes con mutaciones en DGKE que además asocian mutaciones en otros genes previamente relacionados con SHUa, como THBD y C364, lo que sugiere que al menos en algunos pacientes la desregulación del complemento podría modular la presentación y la evolución de la enfermedad en portadores de mutaciones en DGKE.
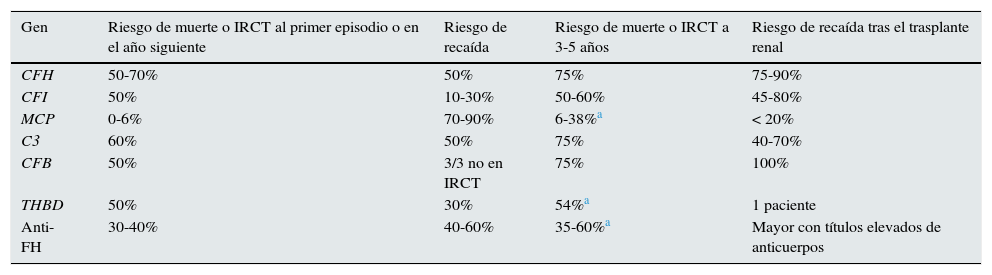
PronósticoLa disponibilidad de Eculizumab ha modificado significativamente el pronóstico de los pacientes con SHUa, enfermedad muy grave en la mayoría de los casos a pesar del tratamiento intensivo con terapia plasmática (TP; tabla 2). Tras un primer episodio de SHUa la mortalidad global era superior al 10% y más de la mitad de los pacientes requerían diálisis y/o presentaban daño renal permanente durante los 12meses siguientes11,12,20. La evolución clínica varía de forma relevante dependiendo de la mutación presente en el paciente. En este sentido, se observaba que la evolución era especialmente sombría en los pacientes con mutaciones en FH y C3, con tasas de mortalidad o de insuficiencia renal crónica terminal (IRCT) durante el año siguiente al primer episodio de SHUa superiores al 50%. Adicionalmente, la mitad de estos pacientes presentaban recaídas de la enfermedad. Las mutaciones en FI, FB y THBD se asociaban también con tasas elevadas de mortalidad/IRCT al año (50%), observándose recaídas de la enfermedad en aproximadamente uno de cada 3 pacientes que superaban el primer episodio de SHUa. Por el contrario, menos del 10% de los pacientes con mutaciones en MCP fallecían o progresaban a IRCT, aunque el riesgo de recaída de estos pacientes era el más elevado y hasta un 90% de ellos sufrían nuevos episodios de SHUa. A los 3-5años del episodio inicial de SHUa, entre el 50 y el 75% de los pacientes con mutaciones en FH, CI, C3, FB o THBD fallecían o presentaban IRCT1.
Evolución clínica de los pacientes con síndrome hemolítico urémico atípico según la alteración del complemento (era pre-Eculizumab)
| Gen | Riesgo de muerte o IRCT al primer episodio o en el año siguiente | Riesgo de recaída | Riesgo de muerte o IRCT a 3-5 años | Riesgo de recaída tras el trasplante renal |
|---|---|---|---|---|
| CFH | 50-70% | 50% | 75% | 75-90% |
| CFI | 50% | 10-30% | 50-60% | 45-80% |
| MCP | 0-6% | 70-90% | 6-38%a | < 20% |
| C3 | 60% | 50% | 75% | 40-70% |
| CFB | 50% | 3/3 no en IRCT | 75% | 100% |
| THBD | 50% | 30% | 54%a | 1 paciente |
| Anti-FH | 30-40% | 40-60% | 35-60%a | Mayor con títulos elevados de anticuerpos |
Anti-FH: anticuerpos anti-factor H del complemento; CFB: gen del factor B del complemento; CFH: gen del factor H del complemento; CFI: gen del factor i del complemento; IRCT: insuficiencia renal crónica terminal; MCP: gen de la proteína cofactor de membrana; THBD: gen de la trombomodulina.
Los resultados del trasplante renal (TR) en los pacientes con IRCT por SHUa se han visto limitados históricamente por el elevado porcentaje de recurrencias de la enfermedad postrasplante (∼50%; tasa de pérdida del injerto: 80-90%65,66), aunque los resultados varían de forma importante en función del tipo de alteración. En una serie de 57 pacientes con SHUa que recibieron un TR se observó que la supervivencia del injerto sin recurrencia a los 5años fue significativamente menor en los pacientes en los que se detectaron mutaciones en los genes que codifican proteínas del complemento vs. los pacientes en los que no se halló alteración genética alguna o solo polimorfismos67. A pesar de ello, es necesario recalcar que el riesgo de recurrencia del SHUa tras el TR en aquellos pacientes en los que no se detectan alteraciones genéticas se considera también elevado68. Las mutaciones en FH son las que asocian con un mayor riesgo de recurrencia o pérdida del injerto tras el TR (75-90%; especialmente las relacionadas con alteraciones del terminal 3′ y la conversión génica entre CFH y CFHR1, resultando en el gen híbrido CFH/CFHR1 [ambas alteraciones afectan la funcionalidad de la región C-terminal de FH]), siendo el riesgo elevado también con las mutaciones en C3 y FI (40-80%; tabla 2)12,42,48,65,67,69-71. Hasta la fecha se han realizado muy pocos trasplantes en pacientes con mutaciones en FB, pero en todos los casos publicados se observó recurrencia del SHUa y pérdida del injerto49,72. En general, los factores plasmáticos del complemento involucrados en el SHUa son de síntesis hepática, por lo que los pacientes con mutaciones en los genes del complemento que codifican estos factores siguen siendo susceptibles al SHUa tras el TR, porque continúan produciendo factores disfuncionales. El MCP por su parte es una proteína transmembrana altamente expresada en el riñón y, consecuentemente, el TR puede corregir este defecto aportando MCP no alterado en el injerto. Más del 80% de los pacientes con mutaciones en MCP no presentan recurrencia del SHUa tras el TR, siendo la tasa de supervivencia a largo plazo similar a la de los pacientes trasplantados por otras causas40,65,66. El riesgo de recurrencia postrasplante en pacientes con mutaciones en THBD62 o con anticuerpos anti-FH no está bien establecido, aunque en estos últimos parece que la recurrencia se relaciona con niveles elevados y persistentes de anticuerpos19,73.
Diagnóstico del síndrome hemolítico urémico atípicoDebido a la evolución rápida y a la gravedad-severidad de la MAT es necesario establecer un diagnóstico diferencial inmediato desde el punto de vista sindrómico que permita iniciar medidas de soporte en las primeras 24-48h de la admisión del paciente. Posteriormente se iniciarán las determinaciones para un diagnostico etiológico de la MAT. En la tabla 3 se muestran los principales procedimientos y pruebas diagnósticas recomendadas para el diagnóstico de MAT, así como las pruebas específicas para el diagnóstico diferencial de los distintos tipos etiológicos de MAT.
Pruebas diagnósticas y procedimientos recomendados en pacientes con microangiopatía trombótica
| Pruebas diagnósticas generales | |
|---|---|
| • Historia clínica completa, incluyendo toma de fármacos, datos de enfermedades sistémicas y antecedentes personales y familiares• Exploración física completa, incluyendo examen de fondo de ojo• Analítica general habitual de sangre y orina• Determinación niveles de haptoglobina• Determinación de los niveles de complemento sérico• Frotis de sangre periférica• Serología de enfermedades sistémicas (ANA, anti-ADN, ANCA, antic-Scl-70, anticentrómero)• Determinación de anticuerpos anticardiolipina y anticoagulante lúpico• Serología para VIH, VHC, VHB, CMV y H1N1• Estudio completo de coagulación, con fibrinógeno, productos de degradación del fibrinógeno y dímeros D• Investigación de infecciones bacterianas causantes de SHU típico y realización de la prueba de la toxina Shiga (si la clínica orienta en este sentido) | |
| Pruebas diagnósticas específicas | |
| • Infección por STEC | • Muestra fecal si diarrea o frotis rectal: cultivo de STEC (MacConkey para E. coli O157:H7); PCR para genes Stx O157:H7 y otros serotipos, y otras características virulentas; ELISA y/o ensayo de cultivo de tejido celular Vero para suero Stx: anticuerpos anti-LPS contra serotipos prevalentes |
| • Infección por neumococos | • Cultivo bacteriano (generalmente) de fluidos corporales estériles; DAT (test de Coombs), prueba viral (respiratoria), radiografía de tórax (derrame pleural asociado de modo característico en casi todos los casos), citoquimia y cultivo de LCR en los casos secundarios a meningitis por neumococo |
| • Alteraciones de la regulación del complemento | • C3, C4 (plasma/suero), AH50• FH, FI, FB (plasma/suero)• Autoanticuerpos anti-FH• Expresión de MCP superficial en leucocitos (leucocitos poli o mononucleares mediante prueba FACS)• Análisis de mutación en el FH, FI, MCP, C3, FB±THBD |
| • Deficiencia ADAMTS13 (adquirida o hereditaria) | • Actividad plasmática de ADAMTS13 o dosis (ELISA)±inhibidor |
| • Metabolismo de la cobalamina: aciduria metilmalónica | • Cromatografía de aminoácidos en plasma/orina (hiperhomocisteinemia, hipometioninemia; homocistinuria); cromatografía de ácidos orgánicos en orina (aciduriametilmalónica)• Análisis de mutación en el gen MMACHC |
ADAMTS13: A Disintegrin And Metalloproteinase with a ThromboSpondin type 1 motif, member 13; ADN: ácido desoxirribonucleico; ANA: anticuerpo antinuclear; ANCA: anticuerpos anticitoplasma de neutrófilos; CMV: citomegalovirus; DAT: prueba de antiglobulina directa; ELISA: ensayo por inmunoabsorción ligado a enzimas; FACS: separador celular activado por fluorescencia; FB: factor B del complemento; FH: factor H del complemento; FI: factor i del complemento; LCR: líquido cefalorraquídeo; MCP: proteína cofactor de membrana; SHU: síndrome hemolítico urémico; STEC: Escherichia coli productor de toxina Shiga; THBD: trombomodulina; VHB: virus de la hepatitis B; VHC: virus de la hepatitis C; VIH: virus de la inmunodeficiencia humana.
Pruebas diagnósticas específicas: adaptado de Loirat et al.2.
En los pacientes con MAT la analítica mostrará la presencia de trombocitopenia (plaquetas <150.000/mm3 o descenso >25% desde el inicio20) y anemia hemolítica microangiopática (hemoglobina <10mg/dl con test de Coombs directo negativo [si bien algunos pacientes con SHU relacionado con neumococo o con H1N1 pueden presentar un test de Coombs directo positivo]25, LDH elevada, descenso de haptoglobina, reticulocitosis y presencia de esquistocitos20,62). En una serie retrospectiva de 50 pacientes con MAT identificada histológicamente se observó que hasta un 44% de ellos presentaban inicialmente cifras normales de plaquetas74. En consecuencia, ante la ausencia de trombocitopenia en pacientes con insuficiencia renal y LDH elevada, el diagnóstico de MAT debería considerarse igualmente. En relación a los esquistocitos, y aunque es posible su detección en la mayoría de pacientes con enfermedad renal, preeclampsia o válvulas mecánicas, un número de esquistocitos >1% es diagnóstico de MAT en ausencia de otra causa conocida75. Por el contrario, la ausencia de esquistocitos no descarta el diagnóstico de MAT.
La observación de niveles elevados de creatinina sérica, filtrado glomerular (FG) bajo o presencia de proteinuria y/o hematuria11,20,69 son indicativos de disfunción renal. En adultos, la realización de una biopsia renal puede ser necesaria ante un fracaso renal agudo para perfilar la etiología, descartar otros procesos y valorar el pronóstico, si bien la indicación y el momento de la biopsia deben valorarse individualmente en los pacientes con sospecha de MAT debido al riesgo de sangrado. En este sentido, en los pacientes con un diagnóstico inequívoco de SHUa (historia familiar positiva, recurrencia, etc.) no se recomienda la realización de una biopsia renal para confirmar el diagnóstico. En pacientes pediátricos, el diagnóstico se basa fundamentalmente en la clínica, aunque en algunas ocasiones será necesaria la biopsia renal (especialmente en los casos de MAT secundaria o en el TR). Se recomienda que los pacientes con sospecha clínica de MAT sean siempre valorados por un nefrólogo, dada la necesidad de una rápida estrategia terapéutica para minimizar el daño renal irreversible.
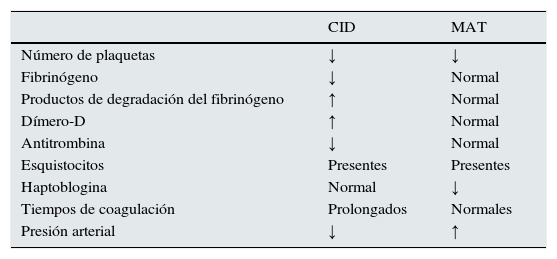
La coagulación intravascular diseminada (CID) es un síndrome que puede asociarse con varios de los principales hallazgos analíticos y clínicos relacionados con MAT. La CID se caracteriza por una activación sistémica de la coagulación, secundaria a diversas situaciones clínicas (sepsis, traumatismos o ciertos tumores), que conlleva la aparición de trombosis y sangrado, con frecuente afectación de la función renal76. La tabla 4 presenta los principales criterios analíticos, basados en el estudio de la coagulación, para el diagnóstico diferencial entre CID y MAT.
Diagnóstico diferencial entre la coagulación intravascular diseminada y la microangiopatía trombótica
| CID | MAT | |
|---|---|---|
| Número de plaquetas | ↓ | ↓ |
| Fibrinógeno | ↓ | Normal |
| Productos de degradación del fibrinógeno | ↑ | Normal |
| Dímero-D | ↑ | Normal |
| Antitrombina | ↓ | Normal |
| Esquistocitos | Presentes | Presentes |
| Haptoblogina | Normal | ↓ |
| Tiempos de coagulación | Prolongados | Normales |
| Presión arterial | ↓ | ↑ |
CID: coagulación intravascular diseminada; MAT: microangiopatía trombótica.
En los pacientes con MAT es necesaria la realización de una historia clínica completa y detallada que incluya antecedentes personales y familiares, identificación de factores desencadenantes (fármacos, infecciones), así como una exhaustiva exploración física. Contrariamente a lo que se consideraba hace años, en la actualidad se admite que los signos y síntomas de los diferentes tipos de MAT no son específicos y no permiten realizar el diagnóstico diferencial entre estas entidades1. Clásicamente, la distinción entre SHU y PTT se basaba en criterios clínicos, considerándose el SHU cuando predominaba la afectación renal y la PTT cuando predominaba la afectación neurológica. Sin embargo, se ha descrito que un 50% de los pacientes con PTT presentan disfunción renal y un 50% de los pacientes con SHUa presentan alteraciones neurológicas37,77. Tampoco la clínica permite diferenciar entre STEC-SHU y SHUa, ya que hasta un 30% de los casos de SHUa se inician tras una gastroenteritis12 o presentan diarrea42 (síntoma característico del STEC-SHU). Por otra parte, la cifra de plaquetas y la severidad de la afectación renal sí que pueden ser orientativas en el diagnóstico diferencial. En general, la PTT cursa con trombocitopenia grave (<20.000/mm3 en el 73% de los pacientes con PTT adquirida78) y moderada afectación renal, mientras que el SHUa suele cursar con trombocitopenia moderada (50-100.000/mm3) y grave afectación renal. Esta regla puede considerarse orientativa, pero la determinación de la actividad de ADAMTS13 y el test de la toxina Shiga resultan esenciales para establecer el diagnóstico diferencial preciso entre PTT, STEC-SHU y SHUa (fig. 4). La detección de la toxina Shiga o el cultivo positivo de STEC en pacientes con MAT es diagnóstico de STEC-SHU28, mientras que el diagnóstico de PTT requiere la demostración de que la actividad plasmática de ADAMTS13 sea <5-10%79,80. En el resto de casos el diagnóstico deberá orientarse hacia SHUa79, siendo necesario realizar pruebas adicionales para descartar MAT secundarias. Las muestras para estudiar deben extraerse antes de que el paciente reciba TP.

Algoritmo para el diagnóstico diferencial de la microangiopatía trombótica primaria.
ADAMTS13: A Disintegrin And Metalloproteinase with a ThromboSpondin type 1 motif, member 13; LDH: lactato deshidrogenasa; PTT: púrputa trombótica trombocitopénica; SHU: síndrome hemolítico urémico; SHUa: síndrome hemolítico urémico atípico; STEC: Escherichia coli productor de toxina Shiga.
* Test de Coombs directo negativo.
** La prueba de la toxina Shiga/STEC está indicada cuando existen antecedentes de afectación digestiva o síntomas gastrointestinales.
*** Excepcionalmente, en algunos pacientes con SHUa la infección por STEC puede ser el desencadenante de la actividad de la enfermedad de base.
El tratamiento del SHUa debe contemplar 2 estrategias distintas: por una parte medidas terapéuticas de soporte encaminadas a controlar las consecuencias del SHUa (fracaso renal agudo, hipertensión arterial, anemia, trombocitopenia, etc.), y el tratamiento específico para frenar y revertir la situación de MAT. En este apartado revisaremos las opciones específicas para controlar el SHUa.
Terapia plasmáticaLas 2 modalidades de TP son la infusión de plasma (IP) y el recambio plasmático (RP). La IP consiste en administrar al paciente plasma fresco congelado viro-inactivado ajeno (fresh frozen plasma [FFP]) aportando reguladores del complemento funcionales81. En el RP se reemplaza el plasma del paciente por FFP, por lo cual no solamente se administran dosis elevadas de proteínas reguladoras del complemento, sino que también se eliminan los inhibidores solubles del complemento disfuncionales endógenos, con menor riesgo de sobrecarga de volumen. Además, en el RP se depuran también los anticuerpos anti-FH y los posibles factores inflamatorios/trombogénicos que participan en el daño endotelial y en la hiperagregación plaquetaria. Clásicamente, el tratamiento de elección recomendado en los episodios de SHUa consistía en la instauración precoz e intensiva de RP a volúmenes elevados en frecuencia variable según la actividad de la enfermedad. Las IP suelen ser ineficaces excepto en los escasos pacientes con déficit completo de FH82 (los niveles circulantes de las proteínas del complemento son normales en la mayor parte de los pacientes). En general, la TP no se considera eficaz en pacientes con mutaciones aisladas de MCP, ya que esta es una proteína no circulante anclada a la membrana celular, observándose que prácticamente la totalidad de estos pacientes remiten tras un episodio de SHUa independientemente del uso de TP12.
Aunque no se dispone de ensayos clínicos prospectivos, la TP ha sido empíricamente el tratamiento de elección para el SHUa durante años tras observarse hace más de 3 décadas que disminuía la mortalidad en pacientes con PTT-SHU. La tabla 5 presenta los resultados del mayor registro internacional de TP en pacientes con SHUa (International Registry of Recurrent and Familial HUS/TTP), que incluye a 273 pacientes diagnosticados entre 1996 y 200712. En dicho registro se observa que con la TP las tasas de recuperación completa hematológica y renal son en general inferiores al 50% (con la excepción de los pacientes con mutaciones en THBD y MCP), siendo dichas tasas especialmente bajas en los pacientes con mutaciones en FH y FI (5 y 12,5%)12. La mortalidad y/o la evolución a IRCT son globalmente elevadas, observándose en 3 de cada 4 pacientes con mutaciones en FI. Algunas publicaciones indican que el RP intensivo precoz es crucial para el rescate del SHUa, y que su mantenimiento puede prevenir la recurrencia de la enfermedad y la IRCT11,81, pero se desconoce el esquema de manejo más eficaz y su impacto a largo plazo sobre la función renal.
Pronóstico de los pacientes con síndrome hemolítico urémico atípico tratados con infusión de plasma o recambio plasmático
| Remisión | Muerte o insuficiencia renal terminal | |
|---|---|---|
| CFH | 63% (completa: 5%; parcial: 58%) | 37% |
| CFI | 25% (completa: 12,5%; parcial: 12,5%) | 75% |
| C3 | 57% (completa: 43%; parcial: 14%) | 43% |
| THBD | 88% (completa: 62%; parcial: 25%) | 13% |
| Anticuerpos anti-FH | 75% (completa: 25%; parcial: 50%) | ND |
| MCP | 97% de los tratados (completa: 90%; parcial: 7%) y 100% no tratados | ND |
Anti-FH: anticuerpos anti-factor H del complemento; CFH: gen del factor H del complemento; CFI: gen del factor i del complemento; MCP: gen de la proteína cofactor de membrana; ND: no disponible; Remisión completa: normalización hematológica y de la función renal; Remisión parcial: normalización hematológica y secuelas renales; THBD: gen de la trombomodulina.
Adaptado de Noris et al.12.
En los pacientes con anticuerpos anti-FH se ha observado que el tratamiento inmunosupresor concomitante a la TP puede mejorar los resultados19,83,84. En estos casos, un título elevado de anticuerpos se correlaciona con un mayor riesgo de recaída y de secuelas renales19. Aunque hacen falta más estudios para establecer de manera conclusiva cómo se generan los anticuerpos anti-FH en los pacientes con SHUa, el hecho de que la inmensa mayoría de estos pacientes presenten una deficiencia completa de FHR1 hace pensar que estos anticuerpos vayan realmente dirigidos contra la proteína FHR1, y que su actividad anti-FH sea una reacción cruzada resultado de la gran homología que existe entre estas 2 proteínas. Esta posibilidad alerta de una posible sensibilización anti-FHR1/anti-FH en individuos homocigotos para la deleción CFHR3-CFHR1 por la exposición a FHR1 exógeno, lo que desaconsejaría la IP en estos individuos.
La aparición de reacciones anafilácticas al FFP, la hipervolemia, la hipertensión arterial, la insuficiencia cardiaca o la hiperproteinemia son complicaciones potenciales de la IP. Las principales complicaciones del RP son la obstrucción de la vía venosa (6%), la hipotensión arterial (5%) y la alergia (4%)85, cuya frecuencia es más elevada en los pacientes pediátricos85. Un estudio realizado en 71 pacientes pediátricos con SHUa (59 tratados con RP) mostró que el 80% de los niños presentaban alguna secuela renal al mes de seguimiento, el 17% mantenían dependencia de diálisis y el 31% presentaron complicaciones relacionadas con el catéter86.
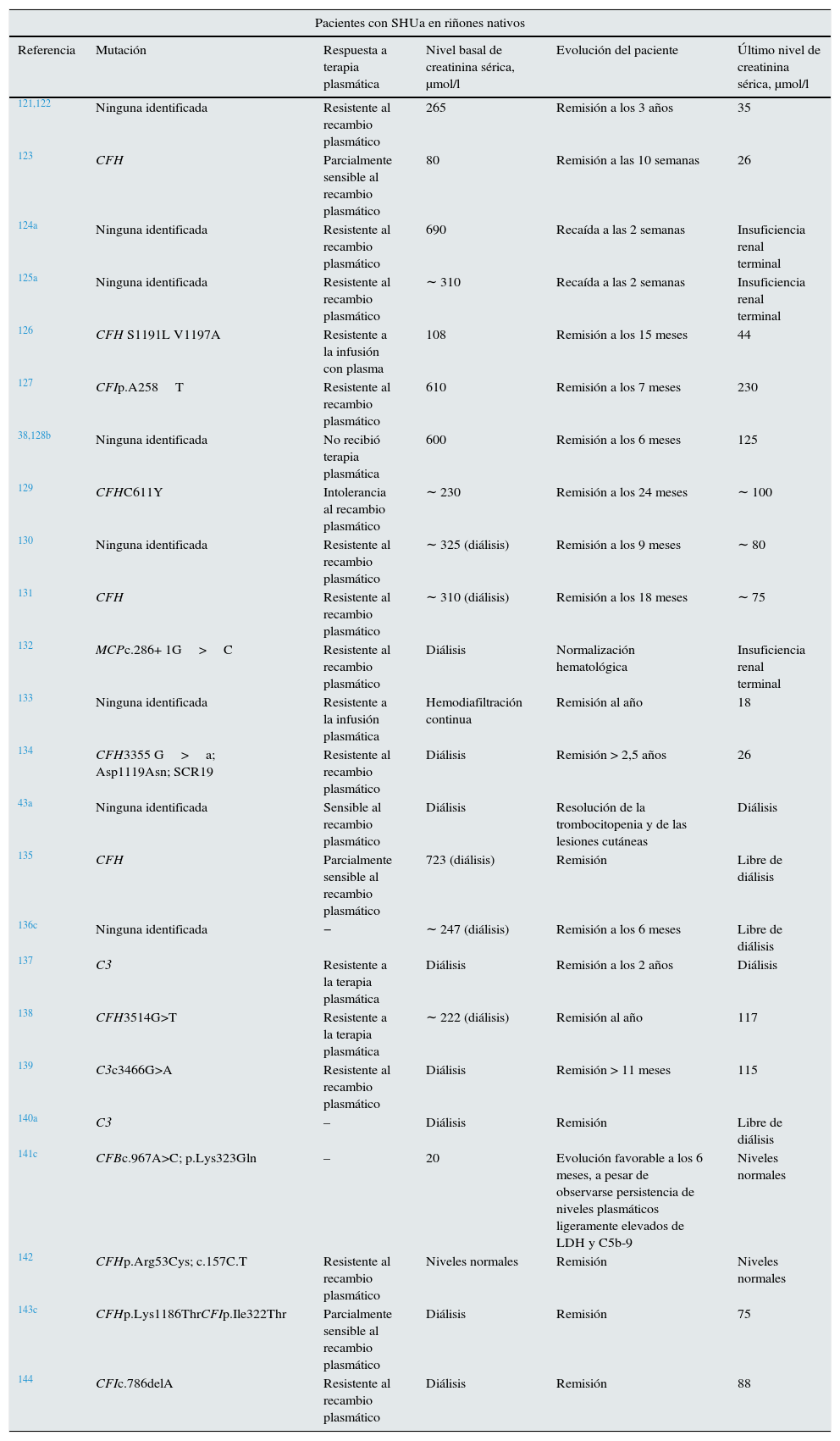
EculizumabEculizumab es un anticuerpo IgG2/4 kappa monoclonal humanizado que se une a la proteína del complemento C5 con gran afinidad, bloqueando su escisión a C5a y C5b e impidiendo la generación del complejo C5b-9 del complemento terminal (complejo de ataque de membrana) (fig. 3)4. En el SHUa, la desregulación de la vía alternativa del complemento conlleva una activación incontrolada de C5 que provoca daño en estructuras propias mediante la formación del complejo de ataque de membrana. El bloqueo de la vía terminal del complemento con Eculizumab reduce rápida y sostenidamente este proceso. En la mayoría de los casos publicados de pacientes con SHUa se ha observado una buena respuesta clínica al fármaco (tabla 6).
Casos publicados de pacientes con síndrome hemolítico urémico atípico que recibieron Eculizumab (actualizado hasta abril de 2014)
| Pacientes con SHUa en riñones nativos | |||||
|---|---|---|---|---|---|
| Referencia | Mutación | Respuesta a terapia plasmática | Nivel basal de creatinina sérica, μmol/l | Evolución del paciente | Último nivel de creatinina sérica, μmol/l |
| 121,122 | Ninguna identificada | Resistente al recambio plasmático | 265 | Remisión a los 3 años | 35 |
| 123 | CFH | Parcialmente sensible al recambio plasmático | 80 | Remisión a las 10 semanas | 26 |
| 124a | Ninguna identificada | Resistente al recambio plasmático | 690 | Recaída a las 2 semanas | Insuficiencia renal terminal |
| 125a | Ninguna identificada | Resistente al recambio plasmático | ∼ 310 | Recaída a las 2 semanas | Insuficiencia renal terminal |
| 126 | CFH S1191L V1197A | Resistente a la infusión con plasma | 108 | Remisión a los 15 meses | 44 |
| 127 | CFIp.A258T | Resistente al recambio plasmático | 610 | Remisión a los 7 meses | 230 |
| 38,128b | Ninguna identificada | No recibió terapia plasmática | 600 | Remisión a los 6 meses | 125 |
| 129 | CFHC611Y | Intolerancia al recambio plasmático | ∼ 230 | Remisión a los 24 meses | ∼ 100 |
| 130 | Ninguna identificada | Resistente al recambio plasmático | ∼ 325 (diálisis) | Remisión a los 9 meses | ∼ 80 |
| 131 | CFH | Resistente al recambio plasmático | ∼ 310 (diálisis) | Remisión a los 18 meses | ∼ 75 |
| 132 | MCPc.286+ 1G>C | Resistente al recambio plasmático | Diálisis | Normalización hematológica | Insuficiencia renal terminal |
| 133 | Ninguna identificada | Resistente a la infusión plasmática | Hemodiafiltración continua | Remisión al año | 18 |
| 134 | CFH3355 G>a; Asp1119Asn; SCR19 | Resistente al recambio plasmático | Diálisis | Remisión > 2,5 años | 26 |
| 43a | Ninguna identificada | Sensible al recambio plasmático | Diálisis | Resolución de la trombocitopenia y de las lesiones cutáneas | Diálisis |
| 135 | CFH | Parcialmente sensible al recambio plasmático | 723 (diálisis) | Remisión | Libre de diálisis |
| 136c | Ninguna identificada | − | ∼ 247 (diálisis) | Remisión a los 6 meses | Libre de diálisis |
| 137 | C3 | Resistente a la terapia plasmática | Diálisis | Remisión a los 2 años | Diálisis |
| 138 | CFH3514G>T | Resistente a la terapia plasmática | ∼ 222 (diálisis) | Remisión al año | 117 |
| 139 | C3c3466G>A | Resistente al recambio plasmático | Diálisis | Remisión > 11 meses | 115 |
| 140a | C3 | – | Diálisis | Remisión | Libre de diálisis |
| 141c | CFBc.967A>C; p.Lys323Gln | – | 20 | Evolución favorable a los 6 meses, a pesar de observarse persistencia de niveles plasmáticos ligeramente elevados de LDH y C5b-9 | Niveles normales |
| 142 | CFHp.Arg53Cys; c.157C.T | Resistente al recambio plasmático | Niveles normales | Remisión | Niveles normales |
| 143c | CFHp.Lys1186ThrCFIp.Ile322Thr | Parcialmente sensible al recambio plasmático | Diálisis | Remisión | 75 |
| 144 | CFIc.786delA | Resistente al recambio plasmático | Diálisis | Remisión | 88 |
| Pacientes trasplantados renales | ||||||
|---|---|---|---|---|---|---|
| Uso preventivo de Eculizumab | ||||||
| Referencia | Mutación | Trasplantes previos (número) | Respuesta a la terapia plasmática | Nivel basal de creatinina sérica, μmol/l | Evolución del paciente | Último nivel de creatinina sérica, μmol/l |
| 107 | CFH W1183C | No | Sensible al recambio plasmático | ∼ 45 | Sin recurrencia | 44 |
| 108 | CFH E1198stop | No | No recibió terapia plasmática | Diálisis | Sin recurrencia | Normal |
| 109 | CFH/CFHR1gen híbrido | No | Sensible al recambio plasmático | Diálisis | Sin recurrencia | 80 |
| 110 | CFH/CFHR1 gen híbrido | No | Sensible al recambio plasmático | Diálisis | Sin recurrencia | Normal |
| 111 | CFH/CFHR1 gen híbrido | No | No recibió terapia plasmática | Diálisis | Sin recurrencia | 79 |
| 112 | CFH c.3497C9T | No | Resistente al recambio plasmático | Diálisis | Sin recurrencia | 76 |
| Uso de Eculizumab para tratar la recurrencia del SHUa tras el trasplante | ||||||
|---|---|---|---|---|---|---|
| 145a | CFH Y475S | Sí (1) | Resistente al recambio plasmático | 132 | Pérdida injerto | NE |
| 146,147 | C3 R570Q | Sí (1) | Sensible al recambio plasmático | 320 | 2 recurrencias cuando hubo retraso en la administración de Eculizumab | 230 |
| 97 | No especificado | No | Resistente al recambio plasmático | 323 | Remisión | 238 |
| 98 | CFH S1191L | Sí (2) | Intolerancia al recambio plasmático | 131 | Remisión | 130 |
| 148a | Ninguna identificada | No | Resistente al recambio plasmático | 415 | Pérdida injerto | NE |
| 42 | CFH | Sí (1) | Resistente al recambio plasmático | 500 | Remisión | 62 |
| 69 | C3 R570W | Sí (2) | Parcialmente sensible al recambio plasmático | 220 | Remisión | 115 |
| 99 | CFH E3514Stop | No | Parcialmente sensible al recambio plasmático | 565 (diálisis) | Remisión | 229 |
| 149 | Ninguna identificada | Sí (1) | Resistente al recambio plasmático | 449 (diálisis) | Recurrencia a los 5 meses de la suspensión de Eculizumab. Pérdida del injerto | NE |
| 43 | CFH | No | Parcialmente sensible a la infusión de plasmad | 220 | Remisión (desaparición de lesiones cutáneas) | 209 |
CFB: gen del factor B del complemento; CFH/CFHR1: gen híbrido del complemento resultante de la conversión entre CFH y CFHR1; CFH: gen del factor H del complemento; CFHR1: gen de la proteína 1 relacionada con el factor H del complemento; CFI: gen del factor i del complemento; LDH: lactato deshidrogenasa; MAT: microangiopatía trombótica; NE: no especificado; SHUa: síndrome hemolítico urémico atípico; SHUa: síndrome hemolítico urémico.
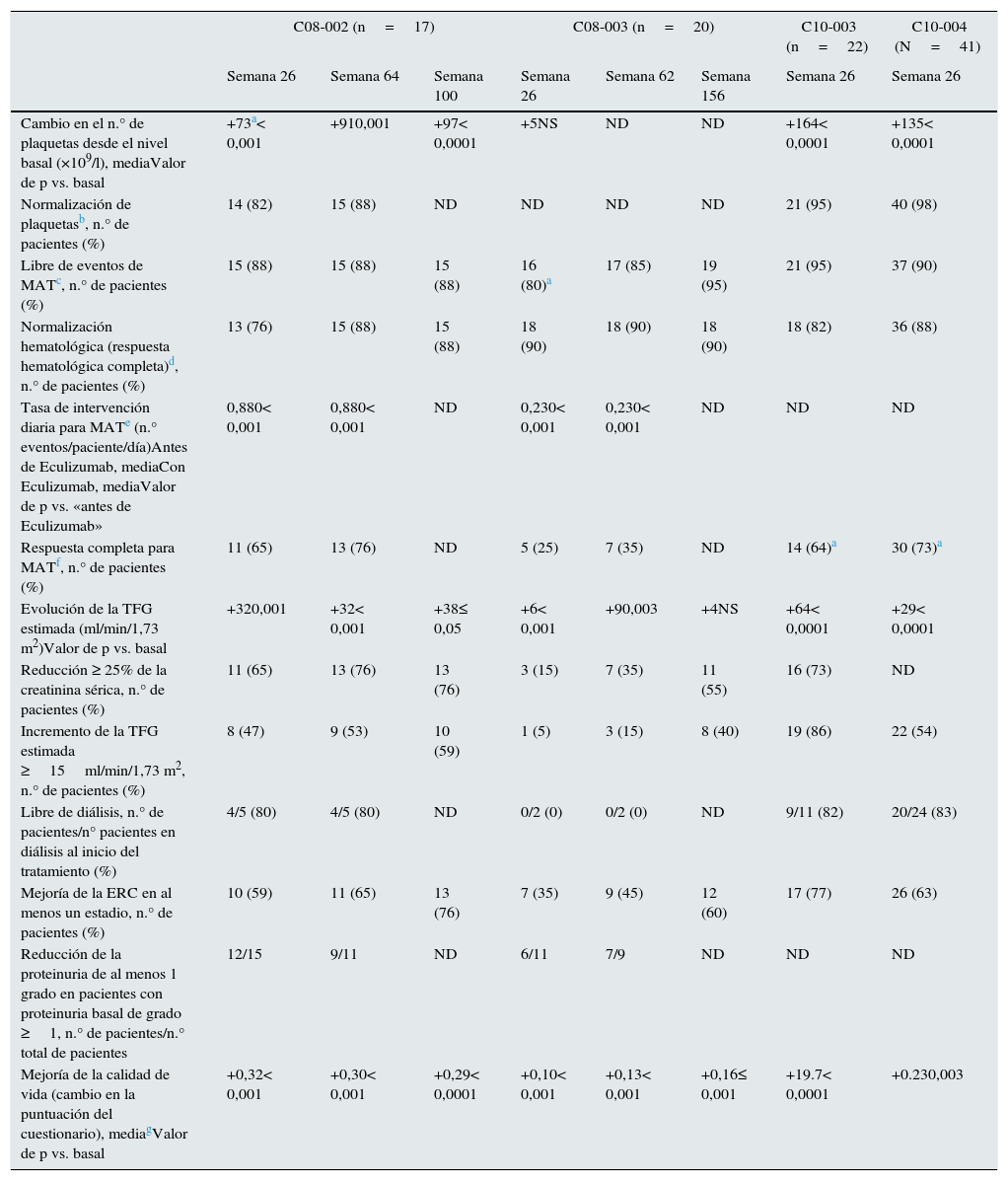
La eficacia y la seguridad de Eculizumab frente al SHUa fueron evaluadas inicialmente en 2 estudios prospectivos multicéntricos de fase 2, en los que 37 pacientes con edad superior a 12años y con enfermedad primaria o recurrente tras el TR recibieron Eculizumab durante 26 semanas, seguidas de periodos de extensión a largo plazo5. En el primer estudio se incluyeron 17 pacientes con SHUa (tiempo medio desde el diagnóstico: 9,7meses) con evidencia de MAT progresiva tras ≥4 sesiones de TP la semana previa a su inclusión (C08-002). En el segundo se incluyeron 20 pacientes (tiempo medio desde el diagnóstico: 48,3meses) en tratamiento con TP (entre una sesión cada 2semanas y 3sesiones por semana) en los que no se observó un descenso >25% en el número de plaquetas durante al menos las 8semanas previas a la primera administración de Eculizumab (C08-003). Se hallaron mutaciones genéticas y/o anticuerpos anti-FH en el 76 y el 70% de los pacientes del primer y segundo estudio, respectivamente. Los objetivos principales en ambos estudios fueron: a)la inhibición de la MAT mediada por el complemento (estudio1: aumento del número de plaquetas; estudio2: paciente libre de eventos de MAT durante ≥12semanas [no disminución de plaquetas >25%, no TP y no inicio de diálisis]), y b)la normalización hematológica (≥2 mediciones consecutivas normales de plaquetas y LDH, separadas un mínimo de 4semanas). Los objetivos secundarios incluyeron los cambios en la tasa de intervenciones diarias para MAT (sesiones de IP o RP, diálisis, o ambas, por paciente/día), la función renal, la calidad de vida, la seguridad y la tolerabilidad. La tabla 7 presenta los principales resultados observados a 26semanas y en los estudios de extensión. Con relación a los objetivos principales, tras 26semanas de tratamiento, en el estudio1 el tratamiento con Eculizumab se asoció con un incremento significativo del número de plaquetas desde el nivel basal (p<0,001) y con una tasa de normalización hematológica del 76%. En el estudio2, el 80% de los pacientes se encontraban libres de episodios de MAT tras 26semanas con Eculizumab y el 90% presentaban normalización hematológica. En cuanto a los objetivos secundarios, el tratamiento con Eculizumab se asoció a las 26semanas con una reducción significativa de la tasa de intervenciones diarias para MAT vs. el nivel basal (p<0,001), así como con una mejoría continuada del FG estimado (+32ml/min/1,73m2 [p=0,001 vs. basal] y +6ml/min/1,73m2 [p<0,001 vs. basal] en los estudios1 y 2, respectivamente), una reducción de la proteinuria (p<0,05) y una reducción de la necesidad de diálisis. Además, cuanto más temprana fue la intervención con Eculizumab en los estudios (menor tiempo de evolución entre la manifestación clínica actual del SHUa y la administración del fármaco), más pronunciada fue la mejoría del FG estimado (p<0,05). Los resultados de los estudios de extensión a uno y 3años demuestran que el tratamiento a largo plazo con Eculizumab se asocia con el mantenimiento o la mejoría progresiva de la respuesta hematológica y de la función renal87,88. Todos estos resultados positivos con Eculizumab se observaron indistintamente en pacientes con o sin alteraciones genéticas y/o anticuerpos anti-FH.
Principales resultados de los estudios prospectivos de Eculizumab en pacientes con SHUa
| C08-002 (n=17) | C08-003 (n=20) | C10-003 (n=22) | C10-004 (N=41) | |||||
|---|---|---|---|---|---|---|---|---|
| Semana 26 | Semana 64 | Semana 100 | Semana 26 | Semana 62 | Semana 156 | Semana 26 | Semana 26 | |
| Cambio en el n.° de plaquetas desde el nivel basal (×109/l), mediaValor de p vs. basal | +73a< 0,001 | +910,001 | +97< 0,0001 | +5NS | ND | ND | +164< 0,0001 | +135< 0,0001 |
| Normalización de plaquetasb, n.° de pacientes (%) | 14 (82) | 15 (88) | ND | ND | ND | ND | 21 (95) | 40 (98) |
| Libre de eventos de MATc, n.° de pacientes (%) | 15 (88) | 15 (88) | 15 (88) | 16 (80)a | 17 (85) | 19 (95) | 21 (95) | 37 (90) |
| Normalización hematológica (respuesta hematológica completa)d, n.° de pacientes (%) | 13 (76) | 15 (88) | 15 (88) | 18 (90) | 18 (90) | 18 (90) | 18 (82) | 36 (88) |
| Tasa de intervención diaria para MATe (n.° eventos/paciente/día)Antes de Eculizumab, mediaCon Eculizumab, mediaValor de p vs. «antes de Eculizumab» | 0,880< 0,001 | 0,880< 0,001 | ND | 0,230< 0,001 | 0,230< 0,001 | ND | ND | ND |
| Respuesta completa para MATf, n.° de pacientes (%) | 11 (65) | 13 (76) | ND | 5 (25) | 7 (35) | ND | 14 (64)a | 30 (73)a |
| Evolución de la TFG estimada (ml/min/1,73 m2)Valor de p vs. basal | +320,001 | +32< 0,001 | +38≤ 0,05 | +6< 0,001 | +90,003 | +4NS | +64< 0,0001 | +29< 0,0001 |
| Reducción ≥ 25% de la creatinina sérica, n.° de pacientes (%) | 11 (65) | 13 (76) | 13 (76) | 3 (15) | 7 (35) | 11 (55) | 16 (73) | ND |
| Incremento de la TFG estimada ≥15ml/min/1,73 m2, n.° de pacientes (%) | 8 (47) | 9 (53) | 10 (59) | 1 (5) | 3 (15) | 8 (40) | 19 (86) | 22 (54) |
| Libre de diálisis, n.° de pacientes/n° pacientes en diálisis al inicio del tratamiento (%) | 4/5 (80) | 4/5 (80) | ND | 0/2 (0) | 0/2 (0) | ND | 9/11 (82) | 20/24 (83) |
| Mejoría de la ERC en al menos un estadio, n.° de pacientes (%) | 10 (59) | 11 (65) | 13 (76) | 7 (35) | 9 (45) | 12 (60) | 17 (77) | 26 (63) |
| Reducción de la proteinuria de al menos 1 grado en pacientes con proteinuria basal de grado ≥1, n.° de pacientes/n.° total de pacientes | 12/15 | 9/11 | ND | 6/11 | 7/9 | ND | ND | ND |
| Mejoría de la calidad de vida (cambio en la puntuación del cuestionario), mediagValor de p vs. basal | +0,32< 0,001 | +0,30< 0,001 | +0,29< 0,0001 | +0,10< 0,001 | +0,13< 0,001 | +0,16≤ 0,001 | +19.7< 0,0001 | +0.230,003 |
EQ-5D: EuroQoL Group 5-Dimension Self-Report Questionnaire; ERC: enfermedad renal crónica; FACIT-F: Functional assessment of chronic illness therapy-fatigue; IP: infusión de plasma; LDH: lactato deshidrogenasa; MAT: microangiopatía trombótica; ND: no disponible; NS: no significativo; RP: recambioplasmático; SHUa: síndrome hemolítico urémico; TGF: tasa de filtrado glomerular-
En general, la tolerabilidad de Eculizumab fue satisfactoria, y únicamente se han reportado 4 casos de efectos adversos graves relacionados con el tratamiento en cada estudio y probablemente en el contexto de la patología de base SHUa (estudio1: hipertensión maligna2, hipertensión severa y bacteriuria asintomática; estudio2: infección por influenza, peritonitis, esclerosis venosa en el punto de infusión y fiebreQ). Debido a su mecanismo de acción Eculizumab incrementa el riesgo de infección por gérmenes encapsulados, especialmente Neisseria meningitidis, por lo que todos los pacientes fueron vacunados contra Neisseria (vacuna tetravalente) 14días antes de iniciar el tratamiento y/o recibieron profilaxis antibiótica, sin observarse ningún caso de meningitis. Tras 3años, solo un paciente ha fallecido por hemorragia digestiva (no relacionada con Eculizumab).
La seguridad y eficacia del uso de Eculizumab durante el embarazo ha sido recientemente evaluada en pacientes con hemaglobinuria paroxística nocturna. Los datos publicados de 61 mujeres con 75 embarazos en tratamiento con Eculizumab sugieren la buena tolerancia de Eculizumab durante el embarazo, con elevadas tasas de supervivencia fetal y materna (96% y 100%, respectivamente). Se detectó la presencia de Eculizumab en 7 de 20 muestras de sangre de cordón umbilical y en ninguna de las 10 muestras de leche materna analizadas89.
Actualmente se encuentran en marcha 2 estudios de fase3, multicéntricos, prospectivos y abiertos, con Eculizumab en pacientes con SHUa. Uno de ellos incluye pacientes adultos (n=41; C10-004)90 y el otro, pacientes pediátricos (n=22; C10-00322)91. A diferencia de los estudios anteriores, los nuevos estudios incluyen mayoritariamente pacientes de diagnóstico reciente de SHUa (73%). En el estudio de adultos, el tiempo medio desde el diagnóstico hasta la inclusión fue de ∼3semanas y el tiempo medio desde el inicio del cuadro clínico hasta la administración de Eculizumab fue de 2semanas (el 15% de los pacientes no recibieron TP de forma previa a Eculizumab)90. Por su parte, el estudio en pacientes pediátricos incluye pacientes con SHUa ≤18años con un tiempo medio desde el diagnóstico hasta la inclusión de ∼2semanas y un tiempo medio desde el inicio del cuadro clínico hasta la administración de Eculizumab de ∼1semana (el 55% no recibió TP)91. Los resultados de estos estudios a 26 semanas confirman las significativas mejorías hematológicas y de función renal observadas en los estudios iniciales, así como los beneficios del uso temprano de Eculizumab (tabla 7)90,91. El perfil de seguridad fue similar, aunque en el estudio de adultos se han observado 2 casos de meningitis meningocócica (5%). Ambas infecciones pudieron ser manejadas correctamente, y uno de los pacientes continuó con Eculizumab. La supervivencia fue del 100% de los pacientes en ambos estudios.
Con el objetivo de valorar el impacto de Eculizumab sobre los biomarcadores relacionados con MAT y daño endotelial en pacientes con SHUa se obtuvieron muestras de sangre de los pacientes adultos incluidos en el estudio C10-004, a nivel basal (antes de iniciar tratamiento con Eculizumab) y en las visitas siguientes, hasta la semana 49-54 de tratamiento92. Se observó que, antes del tratamiento, los pacientes con SHUa (independientemente de la presencia o no de mutaciones relacionadas, de la administración de TP o de los valores hematológicos) presentaban activación del sistema del complemento, junto con parámetros de inflamación, coagulación, activación y daño endotelial y lesión renal. El uso de Eculizumab normalizó los niveles de los biomarcadores relacionados con la activación del complemento y redujo significativamente todos los biomarcadores de inflamación, riesgo trombótico, daño endotelial y orgánico. Eculizumab redujo también los biomarcadores relacionados con la activación de la vía alternativa del complemento y la activación endotelial, si bien los valores de dichos biomarcadores no se normalizaron por completo. Estos hallazgos ponen de manifiesto la persistencia de una desregulación crónica de la activación del complemento en los pacientes con SHUa y de un riesgo continuado de MAT y posible daño orgánico92.
Existe también un estudio retrospectivo de 19 pacientes pediátricos con SHUa tratados con Eculizumab en condiciones de práctica clínica durante 28semanas de media (C09-001)3. En dicho estudio, el 89% de los pacientes normalizaron la cifra de plaquetas y el 68% se mantuvieron libres de episodios de MAT, si bien es necesario tener en cuenta que muchos pacientes se encontraban en remisión hematológica con el tratamiento previo con RP. La tasa de intervenciones para MAT se redujo desde 0,3 por paciente/semana a 0 (p<0,0001). El FG aumentó ≥15ml/min/1,73m2 en el 47% de los pacientes, y en el 50% se eliminó la necesidad de diálisis. Los efectos adversos más frecuentes fueron pirexia (47%), diarrea (32%) e infecciones del tracto respiratorio superior (32%).
Recomendaciones para el manejo del síndrome hemolítico urémico atípicoDesde la aprobación en el 2011 en Europa y España de la indicación de Eculizumab para el tratamiento del SHUa por parte de la EMA y la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS)3, Eculizumab ha permitido mejorar sustancialmente el manejo y el pronóstico de los pacientes con SHUa, ya que la indicación aprobada autoriza su uso en primera línea. A continuación se presentan recomendaciones para el tratamiento del SHUa realizadas por los autores del presente documento a partir de las evidencias disponibles y la experiencia clínica acumulada.
Tratamiento del síndrome hemolítico urémico atípicoConsiderando las dificultades técnicas de la realización de TP en pacientes pediátricos (por el tamaño corporal) y sus potenciales complicaciones, así como por la superioridad de Eculizumab en la recuperación de la función renal (y el consecuente mejor pronóstico), el uso precoz en primera línea de Eculizumab en esta población está especialmente indicado, evitando la realización de RP. En consecuencia, ante la sospecha fundada de SHUa en un paciente pediátrico se recomienda iniciar precozmente la administración de Eculizumab como tratamiento de elección en primera línea (fig. 5)91. En los pacientes adultos con sospecha de SHUa se recomienda iniciar precozmente Eculizumab, previo inicio de RP90. Los RP solo se recomiendan en adultos cuando el diagnóstico no está claro. Únicamente se podría valorar no utilizar Eculizumab en aquellos casos en los que se observe una recuperación completa hematológica y de la función renal tras el inicio de los RP. En este sentido, el Grupo Francés de Estudio del SHUa recomienda transferir al paciente a Eculizumab cuando tras el quinto RP no se logra normalizar las plaquetas o los niveles de LDH, o reducir la creatinina plasmática ≥25%93. Cuando el diagnóstico de SHUa es inequívoco (historia personal o familiar positiva o recurrencia de la enfermedad tras el TR), el tratamiento de elección es Eculizumab. La precocidad en su administración garantiza la reversibilidad del cuadro hematológico y evita la lesión renal.
, debería iniciarse precozmente Eculizumab como tratamiento de elección.")
Tratamiento del síndrome hemolítico urémico atípico.
RP: recambio plasmático; SHUa: síndrome hemolítico urémico atípico.
* Cuando el diagnóstico de SHUa es inequívoco (historia personal o familiar positiva o recurrencia de la enfermedad tras el trasplante renal), debería iniciarse precozmente Eculizumab como tratamiento de elección.
Previamente a la administración de Eculizumab, es necesario vacunar a todos los pacientes frente a N.meningitidis (preferentemente con vacunas tetravalentes conjugadas frente a los serotiposA, C, Y y W135, y serotipoB). En el supuesto de que la administración de Eculizumab no pueda ser diferida hasta obtener la respuesta a la vacunación, podrá iniciarse el tratamiento asociado con cobertura antibiótica contra N.meningitidis e instaurar profilaxis antibiótica3 según el protocolo del hospital. Considerando la frecuencia más elevada de infección invasiva por meningococo en pacientes pediátricos, y la ausencia de protección contra el serotipoB (que es el más prevalente actualmente tras la vacunación sistemática de la población contra otros serotipos), se recomienda mantener a este grupo de edad con profilaxis antibiótica con penicilina o amoxicilina asociada al tratamiento con Eculizumab2, aunque la reciente disponibilidad de la nueva vacuna contra el serotipoB podría modificar estos protocolos de profilaxis. En pacientes adultos se recomienda mantener profilaxis antibiótica mientras se administre Eculizumab según criterio médico y valoración individualizada del paciente. En el caso de pacientes inmunodeprimidos se recomienda valorar el mantenimiento de la profilaxis antibiótica mientras dure el tratamiento con Eculizumab, debido a la menor respuesta a la vacuna que se observa en estos pacientes (especialmente aplicable a la población trasplantada renal). En pacientes pediátricos, la vacunación frente a Haemophilus influenzae y neumococo es también necesaria, así como seguir estrictamente las recomendaciones locales vigentes sobre vacunaciones obligatorias para cada grupo de edad.
En caso de respuesta correcta al Eculizumab, se recomienda continuar el tratamiento de forma indefinida, tal y como recomienda la ficha técnica del fármaco3. Actualmente no es posible realizar recomendaciones sobre la duración más adecuada del tratamiento, aunque a medida que se vayan recogiendo experiencias en el uso del fármaco es posible que en el futuro se pueda definir mejor dicha duración y la estrategia terapéutica.
En algún caso específico se podría valorar la retirada-suspensión y/o la individualización de las dosis de Eculizumab, pero solamente en pacientes de bajo riesgo (con mutación aislada en MCP e historia familiar negativa), siempre de forma individualizada y con un mínimo de 12meses de tratamiento94,95. En los pacientes en los que se retire el fármaco por indicación clínica deberá realizarse una estrecha monitorización durante un mínimo de 12semanas para detectar posibles alteraciones sugestivas de MAT y/o la recidiva del proceso3,96. En estos casos deberá valorarse la reintroducción inmediata de Eculizumab3,96.
En los pacientes con SHUa y fracaso renal agudo severo con necesidad de diálisis se recomienda mantener el tratamiento con Eculizumab un mínimo de 3meses, para valorar la mejoría de la función renal. El incremento progresivo de la diuresis con un buen control tensional son parámetros positivos que orientan hacia un control del proceso de MAT y una mejoría de la lesión renal. La biopsia renal en paciente en diálisis puede ayudar a la toma de decisiones respecto a la continuidad del tratamiento. En caso de fracaso del tratamiento y persistencia de la insuficiencia renal con necesidad de diálisis, se aconseja la suspensión del Eculizumab, excepto en pacientes con manifestaciones sistémicas de la enfermedad, donde debería valorase de manera individual la continuidad del tratamiento.
Cuando se considere la realización de TP en un paciente con SHUa deberá aconsejarse preferiblemente el RP con reposición de FFP (1,5 por volumen de plasma [60-75ml/kg] por sesión para aportar factores de complemento). Deberían realizarse sesiones hasta la normalización de las plaquetas, cese de la hemólisis y mejoría sostenida de la función renal durante varios días. Posteriormente, se aconseja realizar 5 sesiones semanales durante las 2 primeras semanas y 3 sesiones semanales durante las 2semanas siguientes, debiéndose valorar individualmente la continuación del tratamiento con RP2,82.
En los pacientes con SHUa con anticuerpos anti-FH en tratamiento con TP se recomienda el uso concomitante de inmunosupresión para frenar la producción de anticuerpos19,73,83,84. La respuesta al tratamiento en estos casos debería monitorizarse en función de la evolución del título de anticuerpos73.
Las medidas de soporte general son imprescindibles para mantener unas condiciones aceptables del paciente en espera de controlar en proceso de MAT. La hipertensión arterial es frecuente en el contexto de SHUa, recomendándose para su tratamiento el uso de fármacos que bloquean la angiotensinaii (IECA o ARAII). El control de la volemia también resulta esencial por la presencia frecuente de hipervolemia y el riesgo de edema agudo de pulmón. Para el tratamiento de la anemia se valorarán trasfusiones de concentrados de hematíes y/o el uso de factores estimulantes de la eritropoyesis. Las trasfusiones de plaquetas deberán limitarse a plaquetopenias severas (<30.000/mm3) o excepcionalmente en el supuesto de hemorragia grave y/o previo a procedimientos invasivos con riesgo de sangrado, ya que pueden empeorar el fenómeno de MAT. Se deberá además intentar identificar y tratar los posibles agentes desencadenantes del SHUa. Se recomienda que los pacientes pediátricos con SHUa sean transferidos a centros especializados de nefrología pediátrica, con personal experimentado y unidad de cuidados intensivos pediátricos, para garantizar su tratamiento adecuado.
Síndrome hemolítico urémico atípico y trasplanteTratamiento de la recurrencia del síndrome hemolítico urémico atípico en el trasplante renalEn la tabla 8 y en la figura 6 se presentan, respectivamente, los principales criterios clínicos para el diagnóstico diferencial de los distintos tipos de MAT en el TR y un algoritmo de manejo de estas entidades. Ante un paciente trasplantado renal que desarrolle una MAT con antecedentes de episodios de SHUa previos al trasplante, el diagnóstico deberá orientarse hacia una recurrencia de la enfermedad (siendo necesario descartar otras posibles causas).
Diagnóstico diferencial de la MAT en el paciente trasplantado renal
| MAT de novo | Recurrencia del SHUa | |
|---|---|---|
| Antecedente de SHU/MAT | No | Sí |
| Afectación sistémica | No | Frecuente |
| Intensidad del cuadro clínico | Leve | Grave |
| Aparición | Progresiva | Abrupta |
| Intensidad de la MAT hematológica | Baja | Elevada |
| Agentes causantes/desencadenantes | ICN, imTORInfecciones viralesRechazo humoral | No siempre se hallan factores desencadenantes |
| Reversibilidad | Sí | No. Pérdida del injerto |
ICN: inhibidores de la calcineurina; imTOR: inhibidores de la mammalian target of Rapamycin; MAT: microangiopatía trombótica; SHU: síndrome hemolítico urémico; SHUa: síndrome hemolítico urémico atípico.
Adaptado de Zuber et al.68.
; IS: inmunosupresión; MAT: microangiopatía trombótica; RAH: rechazo agudo humoral; RP: recambio plasmático; SHU: síndrome hemolítico urémico; SHUa: síndrome hemolítico urémico atípico; STEC: Escherichia coli productor de toxina Shiga. a La recurrencia del STEC-SHU tras el trasplante renal es una situación muy poco frecuente.")
Tratamiento de la microangiopatía trombótica en el trasplante renal.
CMV: citomegalovirus; ICN: inhibidores de la calcineurina; imTOR: inhibidores de mTOR (mammalian target of Rapamycin); IS: inmunosupresión; MAT: microangiopatía trombótica; RAH: rechazo agudo humoral; RP: recambio plasmático; SHU: síndrome hemolítico urémico; SHUa: síndrome hemolítico urémico atípico; STEC: Escherichia coli productor de toxina Shiga.
a La recurrencia del STEC-SHU tras el trasplante renal es una situación muy poco frecuente.
El tratamiento de la recurrencia del SHUa en un paciente trasplantado renal deberá realizarse en los mismos términos que en el SHUa de riñones nativos mediante la instauración precoz de Eculizumab5,42,43,69,90,91,97,100. Debido al estado de inmunosupresión inducido en los pacientes trasplantados (tratamiento inmunosupresor crónico), se recomienda, además de vacunar frente a la N.meningitidis, valorar mantener la profilaxis antibiótica mientras dure el tratamiento con Eculizumab.
En el estudio inicial de Eculizumab en pacientes con MAT progresiva se observó que la recuperación de la función renal tras la introducción del fármaco era significativamente mejor a largo plazo en los pacientes no trasplantados (con SHUa en riñones nativos) en comparación con los pacientes trasplantados renales5. Probablemente este resultado se relaciona con el hecho de que los pacientes trasplantados que se incluyeron en el estudio recibieron Eculizumab más tardíamente que los pacientes no trasplantados (tiempo medio desde el inicio del cuadro clínico hasta la inclusión de 1,71 y 0,67meses para ambos tipos de pacientes, respectivamente). Esta observación refuerza la necesidad de introducir Eculizumab precozmente ante una recurrencia del SHUa en el TR.
Profilaxis de la recurrencia del síndrome hemolítico urémico atípico tras el trasplante renalEn los últimos años, las perspectivas del TR en los pacientes con IRCT secundaria a SHUa en diálisis han cambiado significativamente, especialmente en los pacientes con alto riesgo de recurrencia de la enfermedad tras el trasplante (pacientes con mutaciones de riesgo y/o con episodios recidivantes de SHUa). La utilización de Eculizumab ha abierto opciones terapéuticas en dichos pacientes, en los que el TR estaba contraindicado por el alto índice de recidiva y el riesgo de pérdida del injerto renal. Actualmente existen 3 opciones terapéuticas para prevenir la recurrencia del SHUa tras el TR: a)la realización de un trasplante combinado hepatorrenal; b)el TR simple con uso de TP profiláctica, y 3)el TR simple con uso de Eculizumab profiláctico.
Hasta la fecha se han publicado más de 20 casos de pacientes con SHUa y mutaciones en genes que codifican factores del complemento de síntesis hepática (FH, FB o FI) en los que se ha practicado un trasplante hepático (aislado o combinado con TR) con el objetivo de evitar las consecuencias del defecto genético y prevenir la recurrencia de la enfermedad. Esta estrategia, combinada con la administración peroperatoria de Eculizumab o de plasma (para eliminar los factores del complemento disfuncionantes durante la cirugía y aportar suficientes factores funcionales hasta la recuperación de la función hepática), ha resultado exitosa en varios casos, observándose una buena función hepática y la ausencia de recurrencia del SHUa en el seguimiento70,101-105. No obstante, debido a la potencial morbimortalidad relacionada con el trasplante hepático2, así como a la dificultad de disponibilidad de órganos y la existencia de alternativas más seguras, se recomienda valorar la indicación del trasplante hepatorrenal en pacientes con SHUa únicamente como segunda opción en casos seleccionados. Por otra parte, no se recomienda el trasplante hepático aislado en pacientes con SHUa y riñones funcionales, ya que los riesgos del tratamiento inmunosupresor crónico superan los relacionados con el tratamiento a largo plazo con Eculizumab68.
El uso de TP profiláctica en los pacientes con IRCT secundaria a SHUa que reciben un TR simple se ha asociado en varias publicaciones con buenos resultados en términos de prevención de la recurrencia de la enfermedad68. A pesar de ello, se han reportado casos de recurrencia del SHUa tras el TR en pacientes de riesgo, incluso con el uso de TP intensiva68,100. Por otra parte, factores como el posible aumento del riesgo de recurrencia de la enfermedad con el progresivo espaciamiento de las sesiones de TP106 o el impacto sobre la calidad de vida del paciente por la administración de TP a largo plazo (relacionado en gran parte con la necesidad de mantener un acceso vascular) limitan la indicación de esta estrategia68.
En los últimos años se han publicado varias experiencias positivas con el uso de Eculizumab profiláctico en pacientes pediátricos con mutación en FH previo a un TR de donante cadáver107-111 (tabla 6), sugiriendo que el TR asociado con el uso preventivo de Eculizumab representa una opción eficiente y bien tolerada en estos pacientes68. Recientemente, Zuber et al.100 publicaron una serie de 9 pacientes tratados de forma profiláctica con Eculizumab para prevenir la recurrencia del SHUa en el post-TR100. Se trata de 6 casos pediátricos y 3 adultos con diferentes mutaciones en la vía alternativa del complemento (5 en FH, uno en C3 y 3 pacientes con genes híbridos resultantes de la recombinación no homóloga entre CFH y CFHR1). Dos pacientes recibieron RP post-TR y posteriormente se transfirieron a Eculizumab, 2 pacientes recibieron Eculizumab desde la semana previa al trasplante (donante vivo no relacionado; urgencia en donante cadáver) y los 5 restantes recibieron Eculizumab desde el postrasplante inmediato. Excepto un caso en que una trombosis temprana llevó a la pérdida del injerto, los 8 casos restantes presentaron una evolución favorable sin recurrencias tras un seguimiento medio de 14,5meses (creatinina media: 71,6±44,8μmol/l). Blasco Pelicano et al.112 han publicado el primer caso en nuestro país de uso profiláctico de Eculizumab en una paciente adulta con mutación en el FH, con una correcta evolución y sin signos de recidiva después de 3años del TR112. En consecuencia, en pacientes con IRCT secundaria a SHUa candidatos a TR se recomienda el uso profiláctico de Eculizumab como primera opción para la prevención de la recurrencia del SHUa tras el trasplante68,93. La figura 7 presenta un algoritmo para la valoración previa y el manejo de los pacientes con SHUa candidatos a TR.
, junto con un cribado para anticuerpos anti-FH. b El TR con donante vivo no relacionado se considerará solo si se dispone de Eculizumab.")
Recomendaciones de manejo de los pacientes con IRCT secundaria SHUa candidatos a TR. Adaptado de Zuber et al.68.
CFB: gen del factor B del complemento; CFH/CFHR1: gen híbrido del complemento resultante de la conversión entre CFH y CFHR1; CFH: gen del factor H del complemento; CFHR1: gen de la proteína 1 relacionada con el factor H del complemento; CFI: gen del factor i del complemento; DGKE: gen de la proteína diacilglicerol quinasa-¿; FH: factor H del complemento; IRCT: insuficiencia renal crónica terminal; MCP: gen de la proteína cofactor de membrana; SHUa: síndrome hemolítico urémico atípico; TR: trasplante renal.
a Determinación de niveles plasmáticos de C3, C4, FH, FI y FB, así como la expresión de MCP en leucocitos periféricos; estudio genético completo para detectar mutaciones del complemento conocidas (y polimorfismos de riesgo), junto con un cribado para anticuerpos anti-FH.
b El TR con donante vivo no relacionado se considerará solo si se dispone de Eculizumab.
Existen también experiencias positivas en la realización de un trasplante hepatorrenal con el uso de Eculizumab profiláctico105, aunque, tal y como se ha comentado, la indicación del trasplante hepatorrenal en pacientes con SHUa debería considerarse como segunda opción en casos seleccionados.
Clásicamente se ha considerado contraindicada la donación de vivo para los pacientes con SHUa debido a las elevadas tasas de recurrencia de la enfermedad y de pérdida del injerto, así como por el riesgo de que el donante tenga mutaciones en el sistema del complemento no detectadas que le condicionen el desarrollo posterior de un SHUa1,66,113. Actualmente, los avances realizados en el campo del diagnóstico genético y la disponibilidad de Eculizumab permiten considerar el trasplante de donante vivo relacionado como una opción válida en pacientes con SHUa. En los donantes deberá realizarse siempre un estudio genético-molecular completo, considerándose aptos para la donación únicamente aquellos casos con mutaciones identificadas en el paciente y ausentes en el donante. Por el contrario, si el donante relacionado y el receptor comparten algún factor genético de susceptibilidad para el SHUa, o bien no se detectan mutaciones ni en el receptor ni el donante, no deberá realizarse el trasplante de vivo con donante genéticamente emparentado93. Adicionalmente, el trasplante con donante vivo solo podrá considerarse si se dispone de Eculizumab.
Con relación a la inmunosupresión, no hay protocolos específicos basados en estudios prospectivos para reducir el riesgo de recidiva post-TR. En general se considera que los inhibidores de la calcineurina114 y los inhibidores de mTOR68,115 han sido relacionados con el desarrollo de MAT post-TR, observándose además un efecto sinérgico con la combinación de ambos fármacos116. En este sentido, se recomienda hacer un uso cauteloso de estos fármacos en los pacientes trasplantados renales por IRCT secundaria a SHUa. Para ello, puede valorarse la instauración de pautas inmunosupresoras basadas en belatacept, según el riesgo inmunológico del paciente, aunque no existen datos concluyentes en la literatura sobre la mejor estrategia inmunosupresora en la población de riesgo.
Conclusiones- •
El SHUa está causado por una desregulación de causa genética o adquirida de la activación de la vía alternativa del sistema del complemento sobre superficies celulares, que condiciona el desarrollo de MAT sistémica. En los últimos años se han caracterizado múltiples mutaciones y polimorfismos en los genes de ciertos factores del complemento que se relacionan con dicha desregulación.
- •
Clínicamente se caracteriza por la tríada anemia hemolítica microangiopática no inmune, trombocitopenia y disfunción renal aguda, asociada con manifestaciones extrarrenales frecuentemente. Antes de la disponibilidad de Eculizumab, el SHUa se asociaba en general con una elevada mortalidad y/o evolución a IRCT, así como con una elevada recurrencia tras el TR.
- •
Ante un cuadro clínico sugestivo de MAT, el diagnóstico deberá orientarse hacia SHUa si la prueba de toxina Shiga/STEC es negativa, la actividad plasmática de ADAMTS13 >5-10% y se descartan formas secundarias de SHU.
- •
Eculizumab es un anticuerpo monoclonal que inhibe la activación del C5 y la formación del complejo de ataque de membrana, responsable del desarrollo de daño en estructuras propias en el SHUa. En estudios prospectivos en pacientes con SHUa, Eculizumab interrumpió eficazmente el proceso de MAT, asociándose a largo plazo con mejoras significativas hematológicas y de la función renal.
- •
Los autores del presente documento recomiendan el uso precoz de Eculizumab para el tratamiento del SHUa en pacientes pediátricos y adultos con sospecha clínica de SHUa en riñones nativos, con recurrencia del SHUa tras el TR o con SHUa de novo post-TR.
- •
Se recomienda el uso profiláctico de Eculizumab para la prevención de la recurrencia del SHUa en pacientes con IRCT secundaria a la enfermedad que reciben un TR de donante vivo o cadavérico.
- •
Se recomienda valorar el uso de Eculizumab en pacientes con MAT secundarias resistentes al tratamiento habitual.
Documento avalado por la Sociedad Española de Trasplante (SET), la Sociedad Española de Nefrología (SEN) y la Asociación Española de Nefrología Pediátrica (AENP).
Financiación y agradecimientosLa actividad investigadora del Dr. Rodríguez de Córdoba está financiada por el Ministerio de Economía y Competitividad (SAF2011-26583), la Comunidad de Madrid (S2010/BMD2316), la Unión Europea (EURenOmics), la Fundación Renal Iñigo Álvarez de Toledo y el CIBER de enfermedades raras.
Los autores desean agradecer a Alexion Pharmaceuticals el soporte logístico para la realización de las reuniones del Grupo Español de Síndrome Hemolítico Urémico Atípico, así como el soporte editorial por parte de Ogilvy Healthworld.
Conflicto de interesesLa Dra. Ariceta, el Dr. Blasco, el Dr. Campistol, el Dr. Praga, el Dr. Rodríguez de Córdoba, el Dr. Valdés, el Dr. Vilalta y la Dra. Román han desarrollado actividades de consultoría y docencia para Alexion Pharmaceuticals. La Dra. Torra ha formado parte de comités de expertos en SHUa esponsorizados por Alexion Pharmaceuticals. El Dr. Espinosa ha participado en estudios clínicos esponsorizados por Alexion Pharmaceuticals. Ninguna de las actividades mencionadas han influido en la elaboración e interpretación de este manuscrito, siendo los autores del mismo los únicos responsables de los contenidos.
El manejo de las formas secundarias de SHU deberá basarse en el tratamiento de la etiología primaria de la enfermedad y en la realización de RP. Tal y como se ha comentado, existe en la literatura un número creciente de casos publicados de pacientes con SHU secundario que muestran buena respuesta a Eculizumab, hecho que sugiere que en ciertos pacientes con SHU secundario el complemento juega un papel importante en el desarrollo de MAT30-34,117-120. En consecuencia, en pacientes seleccionados con SHU secundario resistente a las medidas terapéuticas iniciales puede valorarse la administración de Eculizumab, probablemente temporal, como terapia de rescate. Por otra parte, la detección de mutaciones en genes del complemento en pacientes con SHU secundario implica que la enfermedad subyacente es en realidad un SHUa, por lo que el tratamiento de estos pacientes deberá realizarse en los términos descritos en el apartado «Recomendaciones para el manejo del síndrome hemolítico urémico atípico».
Aunque no es necesario para el diagnóstico clínico del SHUa, se recomienda realizar una investigación del complemento que incluya los niveles plasmáticos de todos los factores y un estudio genético completo de los pacientes afectos. La recogida de muestras debe realizarse previamente al inicio del tratamiento con recambios de plasma y enviarse a un laboratorio de referencia (tabla 9). Teniendo en cuenta que se han encontrado mutaciones del complemento en pacientes con STEC-SHU y SHU secundarios, se recomienda también en estos pacientes la realización de estudios genéticos de forma individualizada para investigar la posible asociación con alteraciones del complemento/SHUa2.
Protocolo de recogida de muestras para la realización de estudios del complemento en pacientes con síndrome hemolítico urémico atípico
| Si las muestras van a enviarse a un laboratorio de referencia, lo mejor es contactar con el laboratorio antes y seguir sus instrucciones sobre las muestras que necesitan para realizar los ensayos |
| Si no se dispone de un laboratorio de referencia y lo que se pretende es disponer de muestras almacenadas para su análisis posterior, las muestras que se obtendrán son: |
| 10ml de sangre con EDTA |
| 10ml de sangre coagulada, para suero |
| En caso de que el paciente sea un niño pequeño y no se le puedan extraer 20ml de sangre, se pueden obtener 2-3ml de cada una de las muestras (6-9ml en total) |
| La sangre con EDTA se debe centrifugar inmediatamente a 3.000rpm durante 10min a 4°C, tras lo cual se recogerá el plasma en un tubo limpio procurando no arrastrar células rojas. Tras etiquetar 5 tubos con el nombre del paciente, la fecha y una indicación de que se trata de plasma EDTA, distribuir el plasma obtenido en esos tubos, congelándolos inmediatamente y manteniéndolos a −80°C |
| Después de retirar el plasma, guardar también el pellet celular congelado a −80°C para una futura extracción de ADN |
| La sangre coagulada durante una hora a temperatura ambiente se debe centrifugar a 3.000rpm durante 10min a 4°C e inmediatamente separar el suero procurando no arrastrar células rojas en un tubo limpio. Tras etiquetar 5 tubos con el nombre del paciente, la fecha y una indicación de que se trata de suero, distribuir el suero obtenido en esos tubos, congelándolos inmediatamente y manteniéndolos a −80°C |
EDTA: ácido etilendiaminotetraacético.
Dado que el diagnóstico genético de los genes del complemento permite estimar de forma individualizada el pronóstico y el riesgo de recurrencia de la enfermedad, se recomienda de modo general. En el caso de que el paciente sea un potencial candidato a TR, el estudio genético es indispensable.
Para la recogida de los datos y la realización de futuros estudios se ha organizado en nuestro país un grupo de trabajo que coordina y asesora online sobre los estudios del complemento en pacientes con SHUa.
Se recomienda la recogida de muestras de los injertos de los pacientes trasplantados renales por SHUa para futuros estudios.
(tabla 10)
Protocolo de recogida y envío de muestras para la determinación de la actividad de ADAMTS13
| Extraer la sangre por punción venosa en 2 tubos de citrato de 5ml |
| Rotular los tubos con nombre y apellidos del paciente, fecha y hora de extracción |
| Centrifugar los tubos durante 7min a 3.500rpm |
| Recoger sobrenadante (plasma) en eppendorfs para hacer alicuotas. Evitar recoger parte del precipitado (altera los resultados). Las muestras no se deben congelar nunca antes de realizar este paso |
| Rotular los nuevos tubos con los mismos datos |
| Congelar los tubos a −20°C y enviarlos al laboratorio de referencia. Enviar también un tubo de EDTA de 5ml (refrigerado) |
ADAMTS13: A Disintegrin And Metalloproteinase with a ThromboSpondin type 1 motif, member 13; EDTA: ácido etilendiaminotetraacético.



