


La nefropatía membranosa (NM) constituye una de las causas más frecuentes de síndrome nefrótico en sujetos adultos y ancianos, mientras que en niños y adolescentes es rara. La incidencia se sitúa en unos 5-10 casos por millón de población y año. Es una enfermedad causada por anticuerpos dirigidos contra diversos antígenos podocitarios causando daño podocitario y el depósito de inmunocomplejos en la cara externa de la membrana basal glomerular. Clásicamente se han distinguido dos grandes subtipos de NM, la primaria o idiopática y la NM secundaria. No obstante, el descubrimiento de la patogenia básica de muchos casos de NM primaria (anticuerpos dirigidos contra diversos antígenos podocitarios) ha hecho que muchos autores pongan en cuestión esta diferenciación. En las NM secundarias el mecanismo patogénico es también causado por el depósito de inmunocomplejos en la misma localización, pero en este caso los antígenos se asocian a enfermedades sistémicas, tumores, infecciones ó fármacos diversos.
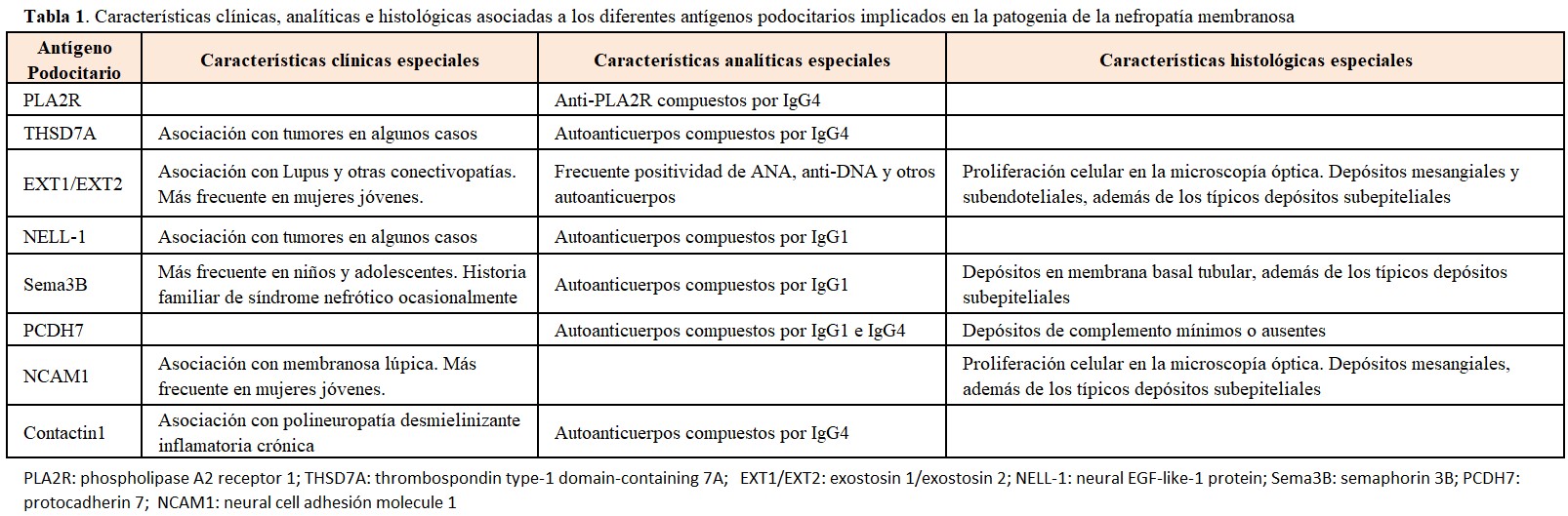
ETIOLOGÍA Y PATOGENIA Nefropatía Membranosa primariaLa búsqueda del antígeno responsable de los casos primarios o idiopáticos ha sido infructuosa durante muchos años. El grupo de Ronco fué el primero en demostrar la participación decisiva de un antígeno podocitario (neutral-endopeptidasa) en un caso de NM infantil [1], aunque los casos de NM primaria atribuibles a este mecanismo son muy raros. Posteriormente se demostró que otra proteína podocitaria, el receptor de la fosfolipasa A2 del tipo M (PLA2R en sus siglas en inglés), constituye el antígeno responsable de un 70-80% de las NM primarias [2]. Los anticuerpos formados contra esta proteína (IgG4 principalmente) atraviesan el capilar glomerular y se unen a la proteína a lo largo de la vertiente externa, o subepitelial, de la pared capilar, formando los típicos depósitos subepiteliales [2]. Posteriormente, a lo largo de los últimos años, se han ido descubriendo otros antígenos podocitarios que ponen en marcha un mecanismo autoinmune similar: thrombospondin type-1 domain-containing 7A (THSD7A), exostosin 1/exostosin 2 (EXT1/EXT2), neural EGF-like-1 protein (NELL-1), semaphorin 3B (Sema3B), protocadherin 7 (PCDH7), neural cell adhesión molecule 1 (NCAM1) y contactin-1 [3][4][5][6][7][8][9][10].
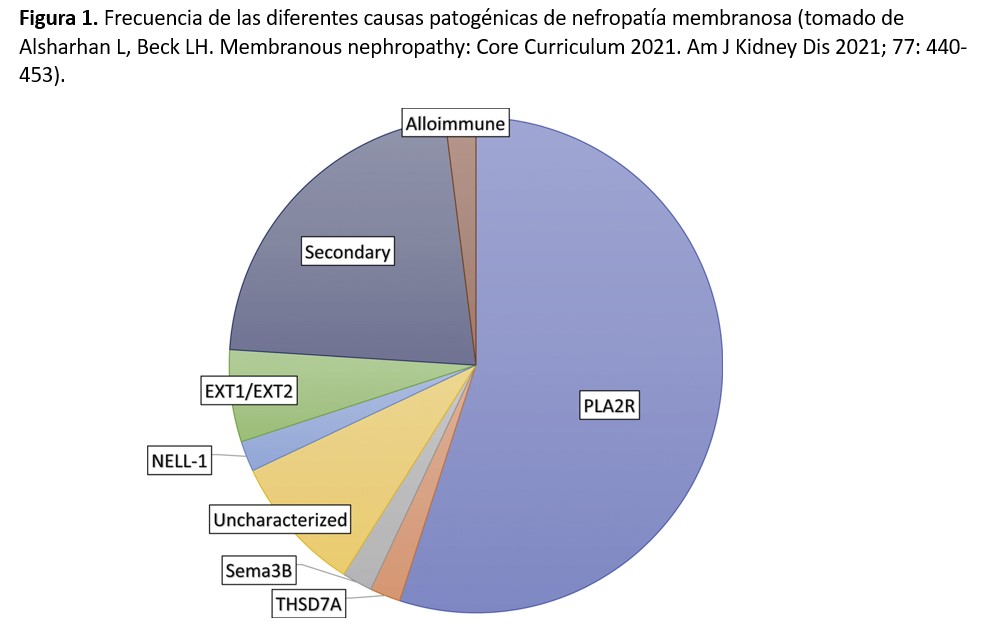
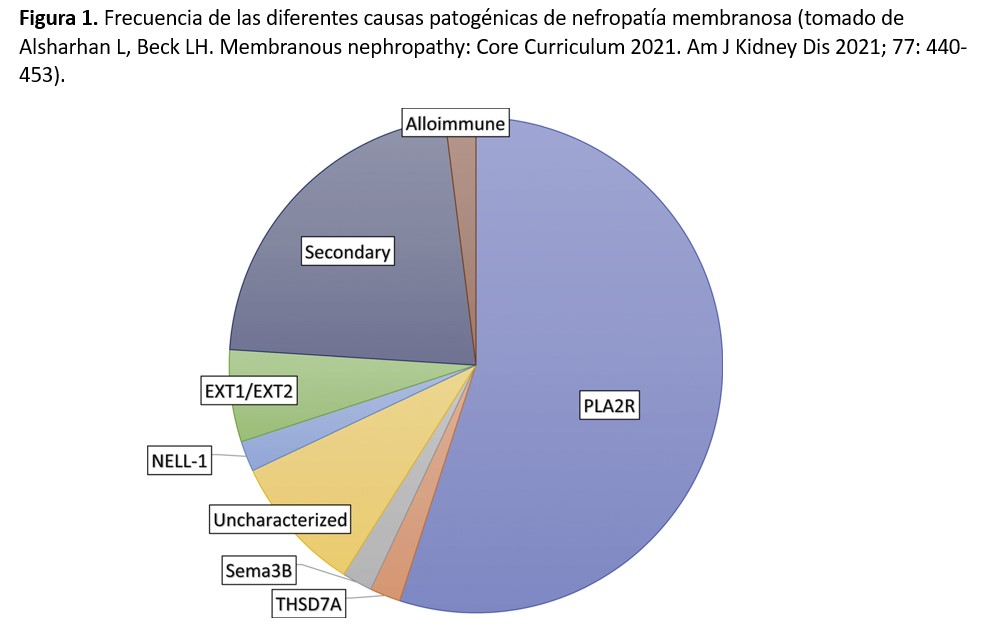
La frecuencia de estos diferentes antígenos aún no está completamente definida, aunque está claro que los casos de NM mediados por anti-PLA2R son con diferencia los más numerosos. En la (Figura 1) se representa el porcentaje aproximado de los diferentes mecanismos patogénicos involucrados en la NM, aunque los más recientemente descubiertos (NCAM1 y contactin-1) no están incluidos en la figura. Otro aspecto importante de estos nuevos antígenos es que varios de ellos se asocian a enfermedades sistémicas o a tumores, además de presentar características clínicas e histológicas peculiares (Tabla 1). La caracterización precisa de estas asociaciones está aún por definir, pero es posible que en un futuro próximo los términos tradicionales de NM primaria o secundaria se cambien por una terminología más precisa asociando cada caso a su etiopatogenia concreta.
A pesar de estos notables avances en la comprensión de las bases patogénicas de la enfermedad, los mecanismos que desencadenan estos procesos de autoinmunidad contra los antígenos podocitarios no están aclarados. Estudios genéticos en cohortes de pacientes han mostrado una base genética, asociada a determinados alelos HLA y a los genes que codifican PLA2R y otros de los antígenos podocitarios descritos. Esta base genética incluye también multitud de variantes genéticas ligadas principalmente a mecanismos inflamatorios, transportes iónicos transcelulares y otros muy diversos y complejos procesos [11][12][13] que predispondrían a la aparición de la enfermedad. Se ha propuesto la utilidad de esta base genética o bien análisis transcriptómicos en tejido renal para el diagnóstico preciso de la enfermedad [12][13].
Se ha sugerido un papel patogénico de aldosa reductasa y superóxido-dismutasa podocitaria en la ampliación del daño podocitario [14] y la participación de antígenos procedentes de la leche de vaca, plantados en la vertiente externa de la pared capilar, se ha demostrado en casos de NM pediátrica [15]. A pesar de este continuo avance en la identificación de antígenos patogénicos en la NM primaria, persisten casos en los que no se logra identificar anticuerpos responsables ni causas secundarias de la enfermedad. El sistema del complemento ejerce un papel patogénico importante en esta entidad. Los anticuerpos, una vez depositados en la pared del capilar glomerular, activan el sistema principalmente a través de la vía de las lectinas. Los factores terminales del complemento (C5-C9) alteran la estructura podocitaria y distorsionan sus diafragmas de hendidura, provocando la aparición de proteinuria masiva [16].
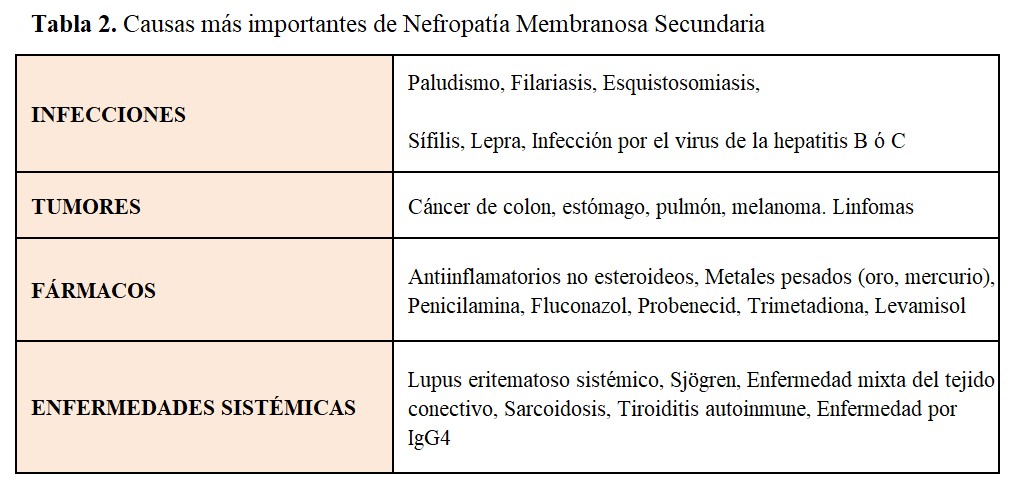
Nefropatías Membranosas secundariasLa NM es un prototipo de enfermedad renal causada por complejos inmunes. Se acepta que los antígenos implicados (de origen infeccioso, farmacológico, tumoral ó de otras diversas procedencias) se depositan primero entre la membrana basal glomerular (MBG) y los podocitos; los anticuerpos específicos generados contra estos antígenos atraviesan la MBG para acoplarse con aquellos, dando lugar a la formación in situ de los complejos inmunes. Existen numerosas entidades que pueden desencadenar una NM secundaria; las más importantes se expresan en la (Tabla 2). La identificación precisa de la causa es esencial, dado que el tratamiento específico de la enfermedad (por ejemplo, resección del tumor, suspensión del fármaco desencadenante ó tratamiento de la infección responsable) conduce en muchos casos a la resolución del síndrome nefrótico. Aunque la determinación de anticuerpos contra THSD7A no se realiza todavía de manera generalizada, su detección obligaría a profundizar en la búsqueda de tumores [17], al igual que la detección de NELL-1 (Tabla 1). Lo mismo ocurre respecto a la asociación de determinados antígenos podocitarios (Exostosin 1/exostosin 2, NCAM1) con enfermedades sistémicas, principalmente lupus eritematoso sistémico.
La determinación de anti-PLA2R ha supuesto un gran avance el diagnóstico diferencial rápido de las NM, para diferenciar las formas primarias de las secundarias. En general, se acepta que la positividad de anti-PLA2R es siempre diagnóstica de NM primaria, aunque coexistan en el paciente otras condiciones (por ejemplo tumores o enfermedades infecciosas) que teóricamente podrían ser las responsables del proceso. Así, se han descrito casos de NM primaria asociados a anti-PLA2R (+) en pacientes con tumores diversos en los que la proteinuria no se modifica tras la extirpación del tumor, reflejando que éste no era la causa de la enfermedad renal.
CUADRO CLÍNICOEn el 80% de los casos la NM se presenta con un síndrome nefrótico completo (proteinuria >3.5 g/24h, hipoalbuminemia, hiperlipidemia) [18][19][20]. La mayoría de los casos, por tanto, son diagnosticados con relativa rapidez, porque el paciente percibe el edema típico del síndrome nefrótico. En el resto de los casos, se detecta proteinuria no nefrótica y el diagnóstico puede retrasarse considerablemente por la ausencia de síntomas. Aunque la presencia de microhematuria es relativamente frecuente, la hematuria macroscópica es muy rara y obliga a descartar la presencia de trombosis de las venas renales o tumores urológicos.
Las manifestaciones clínicas y complicaciones son las de un síndrome nefrótico (edema, hiperlipidemia, hipercoagulabilidad). Las trombosis venosas y, en ocasiones, el tromboembolismo pulmonar consecuencia de la hipercoagulabilidad pueden ser la primera manifestación clínica. El edema suele instaurarse de manera menos abrupta que en el síndrome nefrótico causado por lesiones mínimas clínicas, aunque existe una gran variabilidad.
Analíticamente, los pacientes muestran las anomalías características del síndrome nefrótico (hiperlipidemia, hipoalbuminemia y descenso de proteínas totales). Los niveles de complemento sérico son normales.
En el momento del diagnóstico, la mayoría de casos presenta función renal normal y la tensión arterial suele ser también normal. La aparición de hipertensión arterial suele relacionarse con el desarrollo de insuficiencia renal crónica. Los casos con proteinuria masiva e hipoalbuminemia grave pueden presentar un deterioro progresivo de función renal en los primeros meses de curso clínico. Además, como en todos los tipos de síndrome nefrótico grave, pueden desencadenarse episodios de fracaso renal agudo reversible, por dosis excesivas de diuréticos ó uso de bloqueantes del sistema renina-angiotensina-aldosterona en pacientes con inestabilidad hemodinámica secundaria a la hipoalbuminemia. En los casos más graves, puede detectarse la presencia de glucosuria y otras manifestaciones de tubulopatía [21], lo que probablemente se deba a tubulotoxicidad directa de la proteinuria masiva.
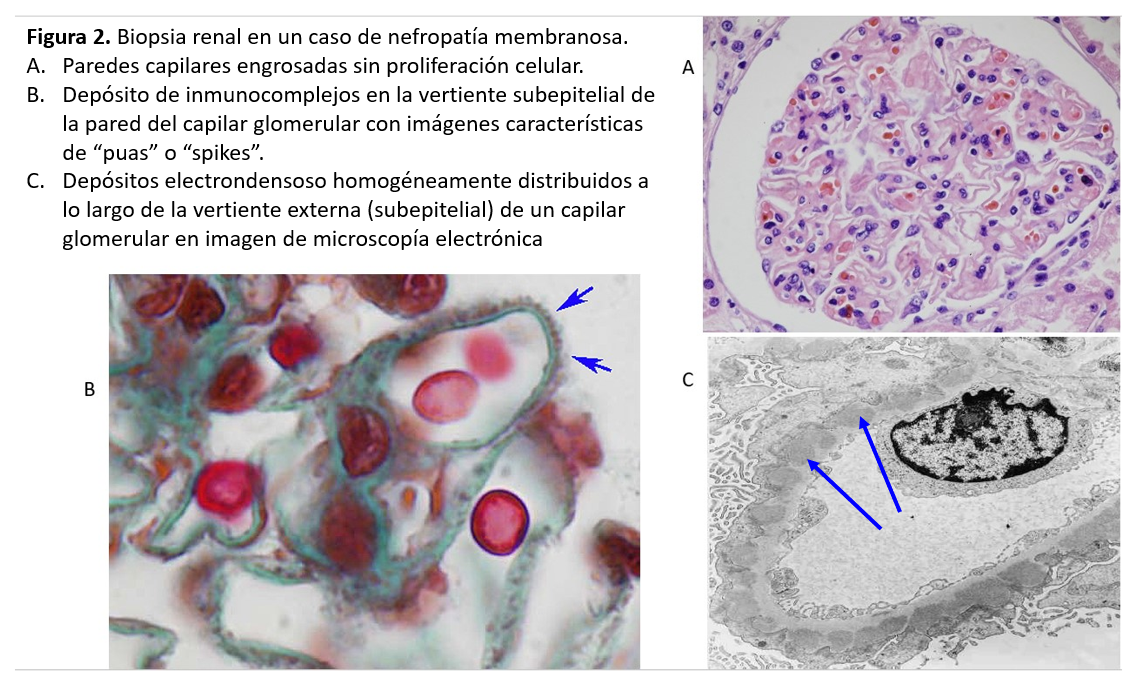
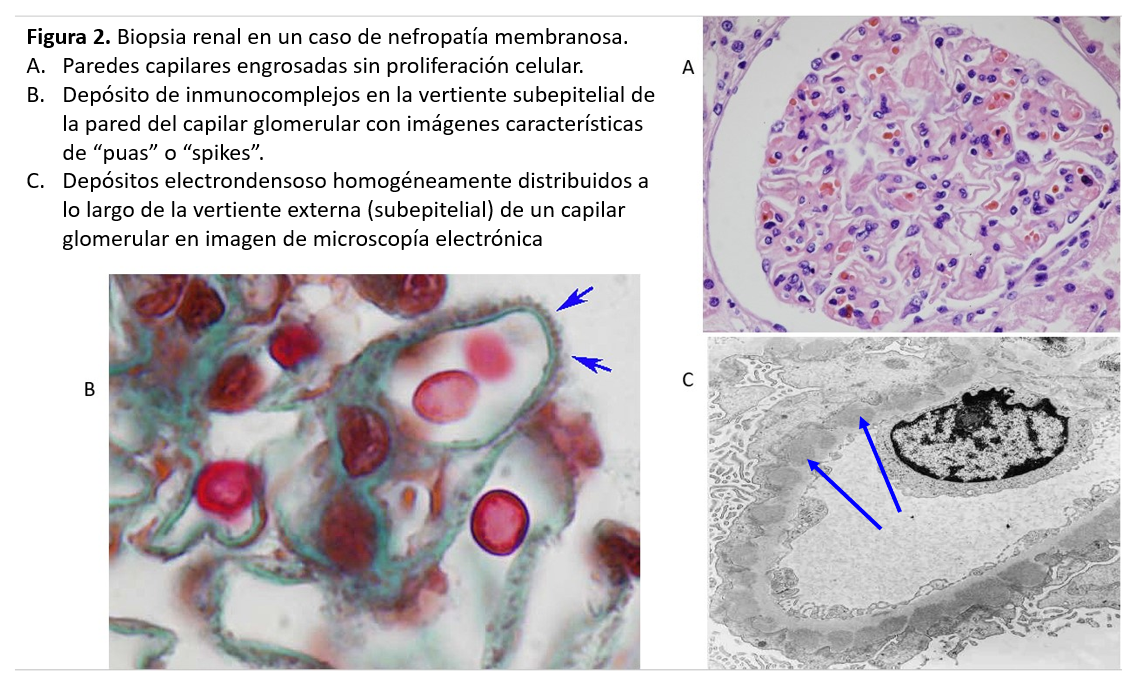
ANATOMIA PATOLÓGICALa NM se caracteriza por un engrosamiento uniforme y difuso de la pared de los capilares glomerulares, sin proliferación celular asociada. El engrosamiento es debido al depósito de complejos inmunes a lo largo de la vertiente externa (subepitelial) de la pared capilar glomerular. La microscopía óptica revela, con las tinciones apropiadas (plata-metenamina), una imágenes muy características de la enfermedad, las llamadas "púas" ó "spikes" en su expresión inglesa (Figura 2). Se trata de prolongaciones espiculares de la membrana basal hacia el exterior que tratan de englobar los depósitos inmunes. Se distinguen cuatro estadios anatomopatológicos de la NM: en el estadio I, se observan los depósitos de inmunocomplejos, pero la pared capilar es aún normal, sin engrosamiento o con un mínimo ensanchamiento difícil de diferenciar de la normalidad óptica. En el estadio II, son ya evidentes el engrosamiento de la pared capilar glomerular y las "púas"o "spikes" en las tinciones con plata. En el estadio III, las prolongaciones de la membrana basal han logrado ya rodear los inmunocomplejos y las paredes capilares muestran un claro engrosamiento y desestructuración. Finalmente, en el estadio IV se observa una esclerosis avanzada, tanto de numerosos glomérulos como del túbulointersticio.
Es importante señalar que los estadios anatomopatológicos tienen una pobre correlación con el pronóstico y con la respuesta al tratamiento, a excepción de la fibrosis túbulointersticial [18][19].
Por otra parte, en el estadio I, la microscopía óptica no permite la diferenciación con otras causas de síndrome nefrótico, como las lesiones mínimas. Es necesaria la inmunofluorescencia, que muestra depósitos de IgG y C3 a lo largo de la pared capilar y la microscopía electrónica, cuyo hallazgo característico es la presencia de depósitos electrón-densos distribuidos homogéneamente por la vertiente subepitelial de todos los glomérulos (Figura 2).
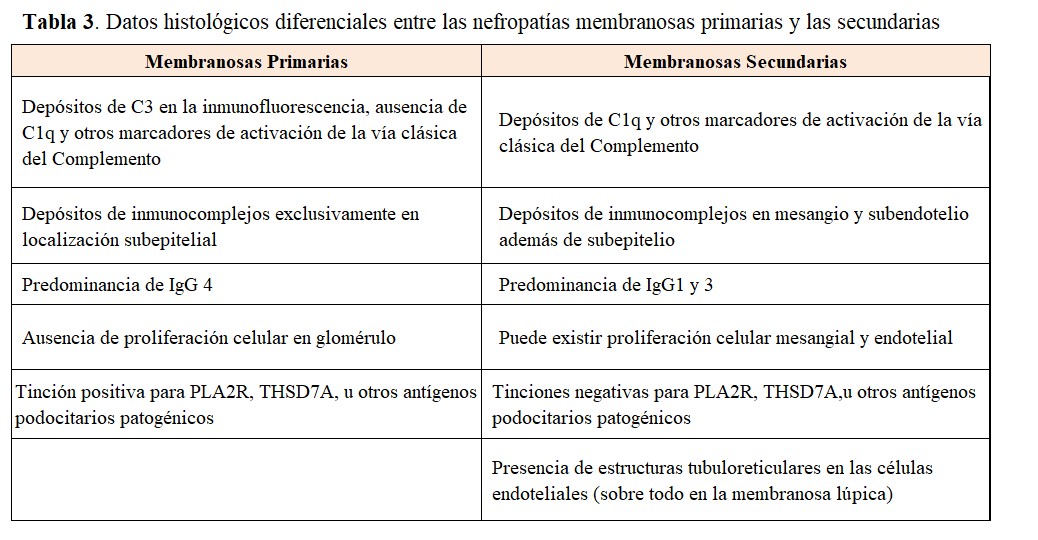
La (Tabla 3) resume los datos histológicos que deben de orientar hacia la sospecha de una NM secundaria [22][23][24], como son la presencia de marcadores de activación de la vía clásica del complemento, la presencia de inmunocomplejos en mesangio o subendotelio, además de los típicos depósitos subepiteliales, y la predominancia de IgG1-3 en lugar de la IgG4 característica de la NM primaria. La presencia de proliferación celular glomerular y los cuerpos tubuloreticulares en células endoteliales son también datos de sospecha de NM secundaria, sobre todo lúpica.
DIAGNÓSTICOLa biopsia renal ha sido imprescindible para establecer el diagnóstico de NM desde las primeras descripciones de la enfermedad. Sin embargo, esta visión está cambiando desde el descubrimiento de los anticuerpos anti-PLA2R como marcadores diagnósticos y pronósticos. Varios estudios han mostrado que en pacientes con proteinuria y síndrome nefrótico el hallazgo de una positividad de anti-PLA2R permite establecer el diagnóstico de NM sin necesidad de realizar una biopsia renal confirmatoria [25][26]. Como decíamos anteriormente, la clasificación de la NM en 4 estadios histológicos tiene escasa relevancia pronóstica y solamente la presencia de una fibrosis túbulointersticial avanzada (que generalmente se asocia a un descenso evidente e irrecuperable del filtrado glomerular) predice una respuesta a los tratamientos inmunosupresores peor. Por tanto, hoy en día, se acepta el que se pueda establecer el diagnóstico de NM en pacientes con clínica compatible (proteinuria con o sin síndrome nefrótico completo) y anti-PLA2R positivos sin hacer biopsia renal. Las excepciones a esta norma [27] serían los pacientes con filtrado glomerular inferior a 60 ml/min/1.73 m2 (para valorar la posible existencia de cambios irreversibles en el parénquima renal y/o excluir posible patologías concomitantes), curso clínico atípico, datos analíticos sugestivos de otras patologías (por ejemplo positividad de ANA, eosinofilia con deterioro rápido de función renal o casos con síndrome nefrótico persistente pese a negativización de anti-PLA2R).
En relación con la importancia de los anti-PLA2R en el diagnóstico de la enfermedad, debe tenerse en cuenta que en algunos enfermos anti-PLA2R positivos no se detectan anticuerpos circulantes en las primeras fases de la enfermedad, por encontrarse todos ellos depositados en el glomérulo (efecto sumidero). En las semanas siguientes, suelen aparecer los anticuerpos en la circulación, pero esto no siempre ocurre. Se ha descrito que los pacientes con tinción positiva para PLA2R en glomérulos pero sin evidencia de los mismos en la circulación tienen un pronóstico mejor.
CURSO CLÍNICOLos pacientes con proteinuria no nefrótica presentan, en su mayoría, una evolución favorable, con función renal estable y sin hipertensión. En algunos casos, la proteinuria puede aumentar hasta el desarrollo de un síndrome nefrótico completo, pero por otra parte, la aparición de remisiones espontáneas es mayor que en los pacientes con síndrome nefrótico. Entre los pacientes con síndrome nefrótico, se pueden separar tres modos diversos de evolución: aparición de remisión espontánea, persistencia del síndrome nefrótico con función renal conservada y persistencia del síndrome nefrótico con deterioro progresivo de función renal.
La aparición de remisiones espontáneas es una característica clave en la NM, observable en un 30-45% de los casos [28][29][30]. Se entiende por remisión espontánea la desaparición del síndrome nefrótico con mantenimiento de la función renal, en ausencia de tratamiento con corticosteroides o cualquier tipo de tratamiento inmunosupresor. Las remisiones se definen como parciales cuando la proteinuria disminuye por debajo del límite nefrótico (3.5 g/24 h) pero continúa siendo superior a 0.3 g/24h (algunos autores prefieren situar el límite en 0.5 g/24h) y completas cuando la proteinuria se estabiliza por debajo de 0.3 g/24h.
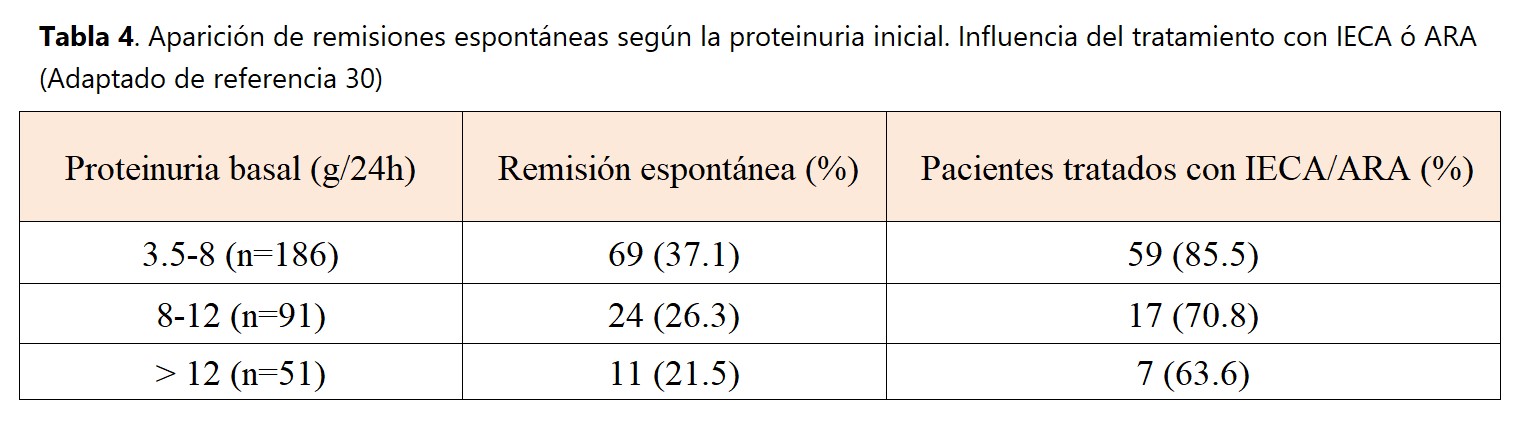
La frecuente aparición de remisiones espontáneas es un hecho conocido desde las primeras descripciones de la NM. Las mujeres y los enfermos con escasa proteinuria tienen una probabilidad mayor de remisión espontánea, así como los pacientes con función renal completamente preservada. En un estudio multicéntrico español que revisó la evolución a largo plazo de más de 300 casos de NM con síndrome nefrótico que no habían sido tratados con corticosteroides u otros inmunosupresores [30], se confirmaron estos datos, pero se observó también que la aparición de remisión espontánea puede verse también en pacientes con graves proteinurias al comienzo de la enfermedad (Tabla 4). Como se observa en dicha tabla, un elevado porcentaje de los enfermos que desarrollaron remisión espontánea recibieron tratamiento con IECA o ARAII. La gran mayoría de remisiones aparecieron en los primeros dos años de la enfermedad y la proteinuria no desapareció abruptamente, sino que mostró un descenso gradual. Los factores que significativamente predijeron una remisión espontánea fueron la función renal y la cuantia de la proteinuria al diagnóstico de la enfermedad, el tratamiento con IECA ó ARAII y la reducción de más de un 50% de la proteinuria basal durante el primer año de evolución. El pronóstico a largo plazo de los enfermos con remisión espontánea completa o parcial fue excelente, con muy escasas recaídas (6%) y una supervivencia renal del 100%.
Por el contrario, un 15-20% de los casos presenta un curso clínico agresivo, con proteinuria masiva y rápido (a lo largo de los primeros 12-24 meses de evolución) declinar de función renal [31][32]. Es importante diferenciar estos casos de evolución agresiva, de aquellos pacientes con fracaso renal agudo por factores reversibles, como puede ser el uso excesivo de diuréticos o factores funcionales sobreañadidos al proceso (hipotensión, deplección de volumen). El pronóstico de estos casos con curso clínico agresivo, en ausencia de tratamiento, es malo [31][32], aunque algunos casos presentan remisiones espontáneas de la proteinuria seguida de una estabilización ó mejoría de la función renal [33].
Finalmente, el resto de pacientes (40-60%) presenta un síndrome nefrótico mantenido, sin desarrollar remisión espontánea ni deterioro de función renal. Aunque esta situación puede persistir años, el pronóstico renal es malo en caso de no remitir el síndrome nefrótico, y el enfermo está expuesto a las complicaciones típicas del síndrome nefrótico: trombosis venosas o un mayor riesgo de accidentes cardiovasculares por la persistente dislipemia.
MARCADORES PRONÓSTICOSLos pacientes con NM necesitan un seguimiento inicial muy estrecho, con revisiones cada 1-2 meses, para seguir la evolución de proteinuria y función renal. Los cambios en estos parámetros van a anunciar la aparición de remisión espontánea o de un curso agresivo, y estas evoluciones dispares suelen definirse en los primeros meses de evolución. La evolución de proteinuria y función renal durante los primeros 6 meses ha sido formulada matemáticamente por el Toronto Registry of Glomerulonephritis para ser aplicada a cada caso individual, con el conocido Toronto Risk Score [34].
Se ha mostrado también que la excreción urinaria elevada de IgG y algunas proteínas de bajo peso molecular, como la α-1 microglobulina y la ß2-microglobulin, es un marcador excelente para predecir el desarrollo de insuficiencia renal, con una sensibilidad del 88% y una especificidad del 91% (35).
Pero sin duda, la determinación seriada de los títulos de anti-PLA2R, en los pacientes positivos para estos anticuerpos, se ha configurado como el marcador más fiable para predecir la evolución y la respuesta al tratamiento [36][37][38][39]. Los títulos elevados de anti-PLA2R se asocian a un peor pronóstico y una peor respuesta al tratamiento. Por otra parte, el tratamiento inmunosupresor disminuye significativamente los títulos de anti-PLA2R, y los pacientes que van a desarrollar una remisión, bien espontánea o inducida por tratamiento muestran descenso de los títulos. Interesantemente, la disminución de anti-PLA2R (remisión inmunológica) precede en varias semanas-meses a la remisión clínica, manifestada por disminución de proteinuria. Los valores elevados de anti-PLA2R pueden predecir también la recidiva de la enfermedad tras el trasplante renal. Se ha propuesto que, en los pacientes positivos, los cambios en el título de anti-PLA2R deberían de guiar el comienzo del tratamiento inmunosupresor o los cambios en el tipo de tratamiento cuando no se aprecia un descenso claro [27][40].
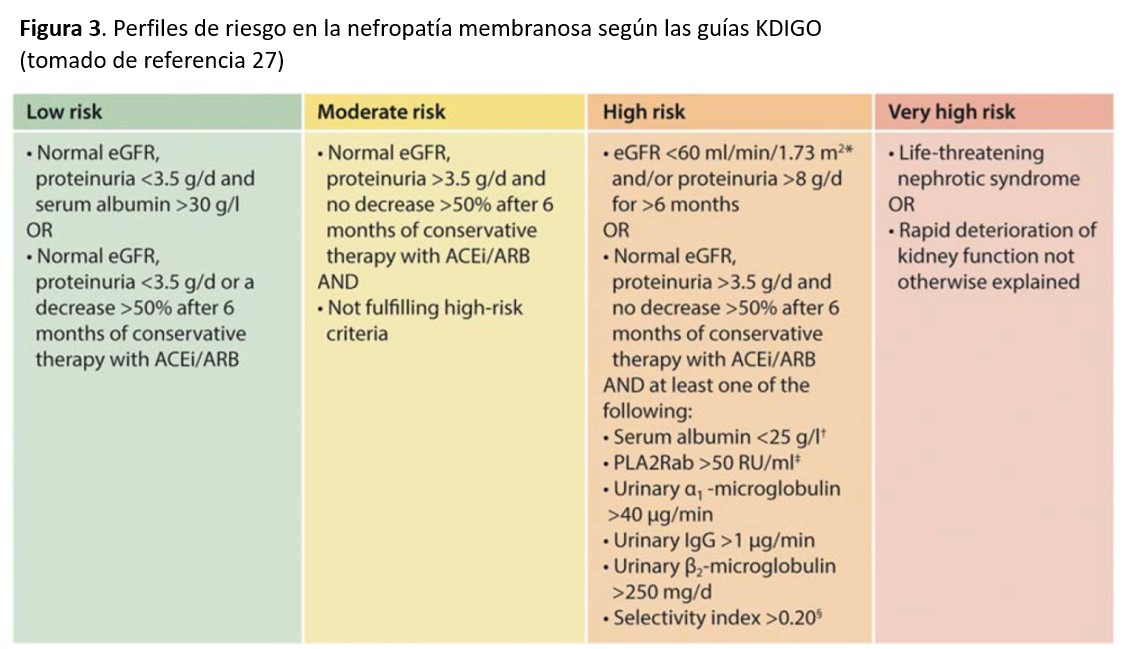
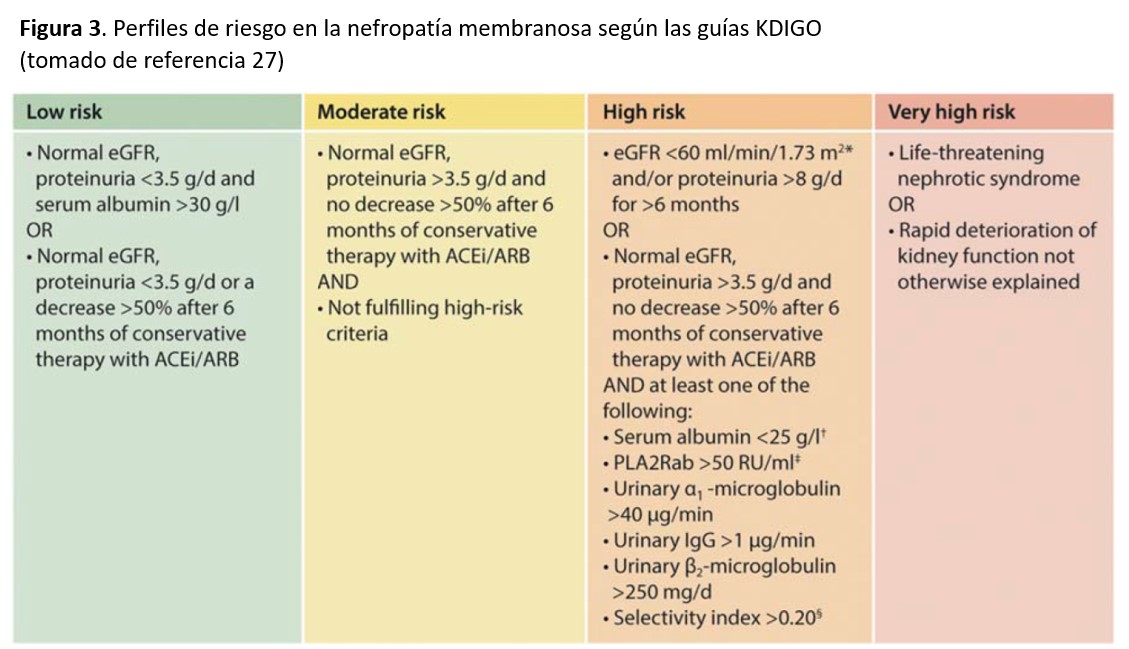
En base a todos estos marcadores pronósticos clínicos, analíticos y serológicos, las guías KDIGO [27] han propuesto cuatro diferentes perfiles de riesgo que deberían de ser considerados a la hora de elegir el tratamiento más adecuado (Figura 3).
TRATAMIENTOEl mejor conocimiento de la historia natural de la NM y estudios terapéuticos prospectivos recientemente publicados han mejorado en los últimos años el enfoque terapéutico de la entidad. Se acepta hoy en día que el tipo de tratamiento debe de adaptarse a las características de cada paciente, teniendo sobre todo en cuenta las variantes evolutivas que comentamos en el apartado de Curso Clínico y los perfiles de riesgo mostrados en la (Figura 3).
Tratamiento conservador. Período de observación sin tratamiento inmunosupresorSe recomienda la instauración de tratamiento inmunosupresor solamente en aquellos pacientes que mantienen proteinuria nefrótica tras un período de observación de al menos 6 meses y siempre que la proteinuria no tenga una clara tendencia a la disminución durante dicho período. En esta fase de observación, debe de instaurarse una serie de medidas conservadoras encaminadas a disminuir los riesgos del síndrome nefrótico, disminuir el edema o facilitar la aparición de remisiones espontáneas como los IECA ó ARAII. De este periodo de observación deberían excluirse aquellos pacientes en los que se observa un progresivo deterioro de función renal (incremento de la creatinina >30% del valor basal durante los primeros 6-12 meses de evolución), los que presentan complicaciones causadas por el síndrome nefrótico (por ejemplo tromboembolismo pulmonar) o los casos con hipoalbuminemia extrema (menor 1.5-2 g/dl) y edema masivo persistente que no responde a combinaciones de diuréticos. Los pacientes con insuficiencia renal crónica avanzada de mucho tiempo de evolución y que muestren importantes lesiones de cronicidad en la biopsia renal tampoco deberían ser candidatos a tratamientos inmunosupresores, dada la improbabilidad de que éstos mejoren el curso clínico del paciente.
Durante el período de observación los pacientes deberían de seguir una dieta sin sal y diuréticos (tiazidas, furosemida, antialdosterónicos) en las dosis y combinaciones requeridas para disminuir el edema y permitir una vida normal. El tratamiento con IECA ó ARAII, además de su indicación en pacientes hipertensos, disminuye la cuantía de la proteinuria y facilita la aparición de remisiones espontáneas, como se comentó anteriormente. Sin embargo, su uso debe de ser cauteloso, particularmente en enfermos sin hipertensión o con un volumen circulante efectivo comprometido por hipoalbuminemia severa.
Un tema controvertido es la indicación de tratamiento anticoagulante. A pesar de que la evidencia se basa solamente en estudios observacionales, las guías KDIGO recomiendan su instauración en pacientes con hipoalbuminemia importante (menor 2 a 2.5 g/dl), sobre todo si presentan además otros factores de riesgo para trombosis: obesidad, antecedentes previos de trombosis o predisposición genética a la misma, insuficiencia cardiaca congestiva, inmovilización ó cirugía abdominal u ortopédica reciente [27]. Sin embargo, debe de valorarse también el riesgo de sangrado en cada paciente, considerando la posible existencia de comorbilidades y los antecedentes personales
Tratamiento de los casos de riesgo bajo y de aquellos que alcanzan remisión parcial espontánea o tras tratamiento inmunosupresor.En estos casos, el mantenimiento de IECA/ARAII para disminuir la cuantía de la proteinuria y mantener las cifras de tensión arterial en los objetivos deseables es primordial. Aunque la evidencia disponible es aún escasa, existen ya datos preliminares que sugieren que los nuevos fármacos renoprotectores y antialbuminúricos (inhibidores de SGLT2, antialdosterónicos de tercera generación como finerenona, anti-endotelina como atrasentan y sparsentan) van a tener un papel muy importante para potenciar la renoprotección ofrecida por el bloqueo del sistema renina-angiotensina y mantener la proteinuria residual en los valores más bajos posibles. Las medidas para disminuir el riesgo cardiovascular (dieta, ejercicio físico, control de tensión, prevención y tratamiento de la obesidad) son también de primordial importancia. La gran mayoría de pacientes requerirán tratamiento hipolipemiante con estatinas.
Tratamiento inmunosupresor en los pacientes de riesgo moderado ó alto.Son varios los tratamientos inmunosupresores que han demostrado su eficacia en el tratamiento de la NM con persistencia del síndrome nefrótico.
Corticosteroides más ciclofosfamida (pauta de Ponticelli)La combinación de corticosteroides y ciclofosfamida o clorambucil, administrados de forma cíclica durante 6 meses (meses 1, 3 y 5, corticosteroides; meses 2, 4 y 6, el agente alquilante) se conoce como pauta o esquema de Ponticelli (Tabla 5). Estudios multicéntricos liderados por este autor han demostrado de manera concluyente la efectividad de este tratamiento, cuando se compara con el manejo exclusivamente conservador. La tasa de remisiones completas o parciales alcanza el 70-80% de casos [41][42][43] y el efecto favorable del tratamiento se ha demostrado con seguimientos prolongados. Los efectos secundarios pueden ser, no obstante, graves. El equipo de Ponticelli demostró que la ciclofosfamida conllevaba un menor número y gravedad de complicaciones, por lo que se ha tendido a abandonar el uso de clorambucil. Medidas preventivas como el trimetroprin-sulfametoxazol para prevenir infección son necesarias.
Regímenes basados en ciclofosfamida diferentes a la pauta de PonticelliLos efectos secundarios de la pauta de Ponticelli, la incomodidad de su administración (choques intravenosos de corticoides al inicio de los meses impares) y la dosis acumulada de ciclofosfamida que puede acarrear han motivado el uso de regímenes diferentes pero también basados en ciclofosfamida. Es importante recordar que la monoterapia con corticoides no es efectiva, según los resultados de un estudio prospectivo [44], aunque es muy probable que potencien la acción de otros fármacos y tengan por sí mismos un efecto favorable al menos transitorio.
Se han utilizado esquemas con administración paralela, no cíclica, de corticoides y ciclofosfamida con resultados en general favorables [31][45]. No obstante, alguno de ellos empleó dosis acumuladas de ciclofosfamida muy elevadas, con la lógica aparición de efectos secundarios graves [46]. Otros estudios han mostrado resultados prometedores administrando uno o dos choques de ciclofosfamida intravenosa en sustitución de la ciclofosfamida oral usada en los meses pares de la pauta de Ponticelli [47][48], con lo que la dosis acumulada del fármaco es sensiblemente inferior. No obstante, estos regímenes alternativos de ciclofosfamida no han sido analizados de manera prospectiva ni controlada.
AnticalcineurínicosEstudios prospectivos randomizados han demostrado que tanto la ciclosporina [49] como el tacrolimus [50][51] son efectivos en la NM, induciendo remisión completa o parcial en más del 70-80% de casos. La dosis inicial habitual de ciclosporina es 3.5-5.0 mg/kg/d, acompañada de esteroides, mientras que la de tacrolimus es 0.05-0.075 mg/kg/d y no necesita ir acompañada de esteroides [51]. Las dosis subsiguientes de ambos fármacos deben de ser ajustadas según niveles sanguíneos. La duración recomenda del tratamiento se sitúa en 12-18 meses y la suspensión debe de ser gradual.
Además de su efecto inmunosupresor, los anticalcineurínicos ejercen un efecto antiproteinúrico directo sobre la estructura del podocito a través de su interacción con la sinaptopodina [52].
La principal limitación en el uso de anticalcineurínicos en la NM es la elevada tasa de recaídas, cercana al 50%, que se observa al suspender el fármaco [49][50]. La administración de rituximab antes de iniciar la reducción de tacrolimus puede disminuir significativamente el número de recaídas [53][54].
En conjunto, como han mostrado estudios observacionales [51], la respuesta a los anticalcineurínicos es tanto mayor cuanto menor sea la proteinuria y mejor la función renal al inicio del tratamiento. Por otra parte, la aparición de recaídas es tanto mayor cuanto mayor sea la proteinuria al inicio de la retirada del fármaco y más rápida sea ésta.
RituximabDiversas series de casos tratados con rituximab, algunas de ellas con un número importante de casos [55][56] muestran que el rituximab induce remisión completa o parcial del síndrome nefrótico en un 50-60% de los casos, con una buena tolerancia y con una tendencia al aumento en el número de remisiones con un seguimiento más prolongado. Las dosis empleadas han oscilado entre 375 mg/m2/semana en cuatro semanas consecutivas o 1 ó 2 dosis de 1 g. No existe una correlación entre la deplección de linfocitos CD20 que el fármaco produce y la aparición de remisiones o recaídas.
El estudio GENRITUX [57] comparó de forma prospectiva y randomizada dos dosis de Rituximab (375 mg/m2 separadas por 7 días) más tratamiento de soporte versus tratamiento de soporte aislado en una serie de 75 pacientes con NM. No se observaron diferencias en el objetivo central del estudio (remisión completa o parcial de la proteinuria a los 6 meses) entre ambos grupos, aunque en los pacientes tratados con rituximab se observó un mayor aumento de albúmina sérica y un descenso más acusado del título de anti-PLA2R. En el seguimiento posterior se comprobó una diferencia significativa, con un significativo mayor número de remisiones entre los que recibieron rituximab. En conjunto, estos datos muestran la eficacia del rituximab en la NM, aunque su efecto parece ser relativamente lento.
MicofenolatoEstudios observacionales sugirieron la eficacia del micofenolato en la NM [58]. Sin embargo, el único estudio controlado publicado hasta la fecha [59] no demostró que los enfermos tratados tuvieran un menor número de remisiones que los controles tratados conservadoramente. Otros estudios observacionales sugieren que el micofenolato puede jugar un papel beneficioso en casos resistentes a otras terapias o que presentan ya grados diversos, no avanzados, de insuficiencia renal [60], pero se requieren más estudios con este tipo de pacientes.
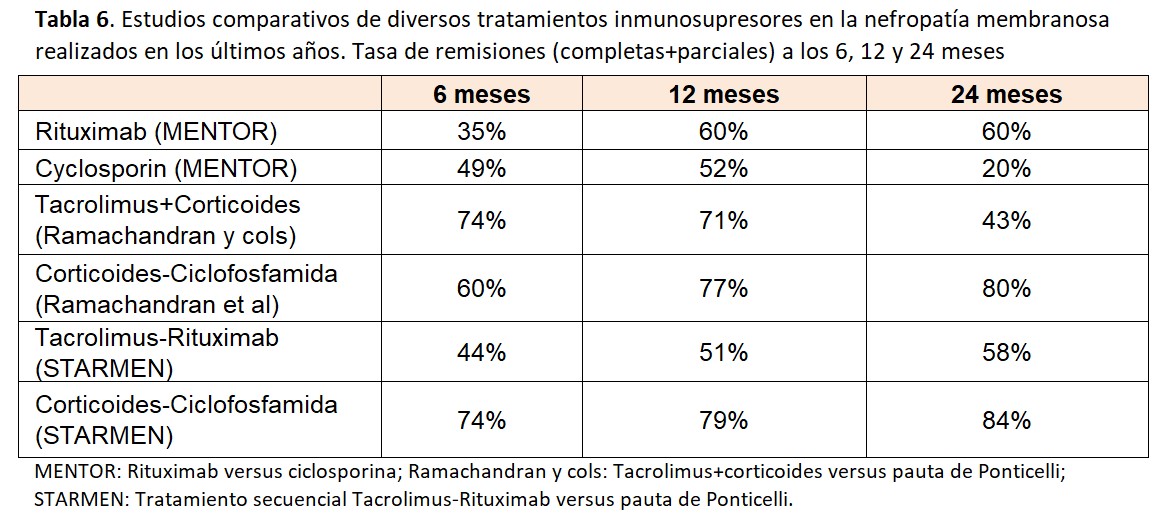
Estudios prospectivos comparativos. Combinaciones de fármacos inmunosupresoresEn los últimos años se han publicado estudios prospectivos controlados que han comparado diversas pautas de tratamientos inmunosupresores (Tabla 6). En el estudio MENTOR [61] se compararon dos dosis de 1 g de Rituximab, separadas por 15 días (con una repetición de la misma dosis a los 6 meses si no existía remisión completa) con ciclosporina (3.5 mg/kg/dia durante 12 meses, con retirada entre los meses 12 y 14). A los 12 meses, no se observaron diferencias significativas en la tasa de remisiones (60% y 52% respectivamente) pero a los 24 meses, estos porcentajes eran claramente favorables para rituximab: 60% versus solamente un 20% con ciclosporina. Esta diferencia se debió al gran número de recaídas en el grupo de ciclosporina, sin duda relacionadas con la rápida retirada de este fármaco.
En un estudio multicéntrico realizado en la India [62] se comparó la pauta clásica de Ponticelli con un tratamiento combinado con corticoides y tacrolimus administrado durante 12 meses. Como se observa en la (Tabla 6), no hubo diferencias en el primer año de tratamiento (77% versus 71% respectivamente) pero a los 24 meses, el número de remisiones era significativamente mayor en los pacientes tratados con la pauta de Ponticelli: 80% versus 43%, observándose también un importante número de recaídas tras la suspensión del tacrolimus. En el estudio STARMEN [54] se comparó la pauta de Ponticelli con un tratamiento secuencial con tacrolimus (dosis completa durante 6 meses y 3 meses de retirada gradual) con una dosis única de 1 gr de rituximab administrado en el 6º mes, al inicio de la disminución de tacrolimus. La tasa de remisiones fue significativamente mayor en el grupo de Ponticelli, alcanzando un 84% a los 24 meses (con un 60% de remisiones completas). Aunque el porcentaje de remisiones fue inferior en el grupo tacrolimus-rituximab, es destacable que alcanzaba un 58% a los 24 meses y que el número de recaídas fue muy bajo, sugiriendo de nuevo la eficacia del rituximab para prevenir las recaídas tras la retirada de los anticalcineurínicos. Finalmente, el estudio piloto RY-CYCLO (63) comparó la pauta de Ponticelli con dos dosis de 1 gr de Rituximab (días 1 y 15). A los 12 meses, la tasa de remisiones completas (objetivo primario del estudio) fue mayor en el grupo de Ponticelli, pero sin alcanzar significación estadística.
Los estudios anteriores incluyeron pacientes con síndrome nefrótico mantenido y función renal estable. Se han realizado muy pocos estudios controlados en NM con deterioro progresivo de función renal. Un estudio prospectivo controlado [64] comparó la pauta de Ponticelli con la ciclosporina y con el tratamiento conservador: la pauta de Ponticelli fue significativamente superior a los otros dos grupos.
Finalmente, se debe considerar la posibilidad de combinar rituximab y ciclofosfamida para aumentar las posibilidades de ambos fármacos. Un estudio recientemente publicado, prospectivo aunque no radomizado, mostró los resultados de una combinación de corticoides (60 mg de prednisona oral con una rápida recucción posterior a lo largo de 6 meses), ciclofosfamida oral (2.5 mg/Kg/día durante 1 semana, 1.5 mg/kg/dia durante 7 semanas más) y rituximab (1 gr IV los días 1 y 15 y posteriormente administraciones de 1 gr cada 4 meses hasta los 24 meses) en una cohorte numerosa de pacientes. Todos ellos alcanzaron remisión del síndrome nefrótico en seguimientos prolongados, la mayoría completas [65].
Tratamiento personalizado de la nefropatía membranosa. Objetivos del tratamientoCon lo dicho anteriormente, hoy en día se recomienda un abordaje terapéutico que considere todas las características del paciente y su perfil de riesgo. Como se resume en la (Figura 4), el tratamiento conservador es el preferible en los casos de riesgo bajo, mientras que el rituximab en monoterapia o combinado con tacrolimus probablemente sean las terapias más adecuadas para los casos de riesgo moderado. Sin embargo en los casos de perfil de alto riesgo los regímenes basados en ciclofosfamida son hoy en día los más eficaces, aunque se requieren más estudios para precisar la validez de regímenes diferentes al de Ponticelli, con administraciones más flexibles de ciclofosfamida, dosis acumuladas menores y combinaciones personalizadas con corticoides y rituximab.
En cuanto a los objetivos del tratamiento, es importante recalcar que, mientras que en otras entidades como la Nefropatía IgA, el pronóstico de pacientes con proteinuria >1g/24h es desfavorable, en la NM existe una tolerancia mayor a cuantías superiores de proteinuria [66]. La inducción de remisión parcial (proteinuria menor 3.5 g/24h) se asocia a una supervivencia renal significativamente superior a la no remisión [67], y el mantenimiento de proteinurias entre 1-3.5 g/24h a lo largo del seguimiento no se asocia al deterioro de función renal observable en otras nefropatías. Por tanto, aunque el objetivo óptimo sería la consecución de una remisión completa, la obtención de remisión parcial puede considerarse satisfactoria.
NEFROPATÍA MEMBRANOSA Y TRASPLANTE RENALLa NM en el trasplante renal puede ser una recurrencia de la enfermedad original o bien tratarse de una NM de novo. La patogenia de ambos procesos es diferente: en las NM recurrentes está bien establecido el papel que juegan los anti-PLA2R y probablemente los nuevos antígenos podocitarios recientemente descritos. De hecho, un título alto de anti-PLA2R debe desaconsejar la realización del trasplante, debiendo esperarse a que dicho título disminuya o desaparezca espontáneamente o con tratamiento. Por el contrario, las NM de novo suelen ser anti-PLA2R negativas y su patogenia es más oscura, habiéndose implicados rechazos previos o infecciones como agentes desencadenantes [68][69][70].
Respecto al tratamiento, la escasa evidencia disponible apunta al rituximab como tratamiento de elección en la gran mayoría de casos [69][70], teniendo en cuenta que los pacientes reciben ya tratamiento inmunosupresor (micofenolato, tacrolimus) y que la ciclofosfamida puede generar efectos secundarios importantes en pacientes ya inmunosuprimidos.





{kind=link}
{kind=link}
{kind=link}
{kind=link}