




La hemoglobina y la mioglobina son hemoproteínas que juegan un papel fundamental en el organismo ya que participan en el transporte de oxígeno. Sin embargo, debido a su estructura química, estas moléculas pueden ejercer efectos deletéreos cuando se liberan al torrente sanguíneo de forma masiva, como sucede en determinadas condiciones patológicas asociadas a rabdomiólisis o hemólisis intravascular. Una vez en el plasma, estas hemoproteínas se pueden filtrar y acumular en el riñón, donde resultan citotóxicas, principalmente para el epitelio tubular, e inducen fracaso renal agudo y enfermedad renal crónica. En la presente revisión analizaremos los distintos contextos patológicos que provocan la acumulación renal de estas hemoproteínas, su relación con la pérdida de función renal a corto y largo plazo, los mecanismos fisiopatólogicos responsables de sus efectos adversos y los sistemas de defensa que contrarrestan tales acciones. Por último, describiremos los distintos tratamientos utilizados actualmente y mostraremos nuevas opciones terapéuticas basadas en la identificación de nuevas dianas celulares y moleculares, prestando especial atención a los diversos ensayos clínicos que se encuentran en marcha en la actualidad.
Haemoglobin and myoglobin are haem proteins that play a key role as they help transport oxygen around the body. However, because of their chemical structure, these molecules can exert harmful effects when they are released massively into the bloodstream, as reported in certain pathological conditions associated with rhabdomyolysis or intravascular haemolysis. Once in the plasma, these haem proteins can be filtered and can accumulate in the kidney, where they become cytotoxic, particularly for the tubular epithelium, inducing acute kidney failure and chronic kidney disease. In this review, we will analyse the different pathological contexts that lead to the renal accumulation of these haem proteins, their relation to both acute and chronic loss of renal function, the pathophysiological mechanisms that cause adverse effects and the defence systems that counteract such actions. Finally, we will describe the different treatments currently used and present new therapeutic options based on the identification of new cellular and molecular targets, with particular emphasis on the numerous clinical trials that are currently ongoing.
La hemoglobina (Hb) y la mioglobina (Mb) son hemoproteínas que juegan un papel fundamental en la homeostasis del organismo, al oxigenar los tejidos y participar en la regulación del pH sanguíneo. La Hb tiene un peso molecular de 64,5kDa y está formada por 4cadenas polipeptídicas denominadas globinas1. Cada globina contiene un grupo hemo con un átomo de hierro en su interior, el cual es responsable de sus propiedades funcionales. La Mb es una proteína más pequeña, con un peso molecular de 17kDa y está formada por una sola globina. En condiciones fisiológicas, tanto la Hb como la Mb se encuentran en el interior de los eritrocitos y de las células del músculo, respectivamente. Sin embargo, en determinadas condiciones patológicas, estas moléculas se liberan al torrente sanguíneo y pueden filtrarse y acumularse en el riñón, donde resultan citotóxicas, principalmente para el epitelio tubular proximal. De hecho, la acumulación renal de hemoproteínas puede inducir tanto fracaso renal agudo (FRA) como enfermedad renal crónica (ERC). En los últimos años se han identificado nuevos mecanismos implicados en el daño renal asociado a estas moléculas, los cuales han servido para desarrollar tratamientos experimentales que ya han dado buenos resultados en estudios publicados recientemente o en ensayos clínicos que se encuentran en marcha, tal y como describiremos más adelante en profundidad.
Origen de la acumulación renal de hemoproteínasLa Mb se acumula en el riñón como consecuencia de un daño muscular severo (rabdomiólisis), mientras que la Hb lo hace a partir de la hemólisis intravascular de glóbulos rojos o de la rotura de hematíes que atraviesan la membrana de filtración glomerular en enfermedades glomerulares hematúricas como la nefropatía IgA (NIgA), lupus o el síndrome Alport, entre otras. En la presente revisión nos centraremos en la mioglobinuria y hemoglobinuria por las limitaciones de espacio.
MioglobinuriaLa mioglobinuria es la presencia de Mb en orina, cuya causa principal es la rabdomiólisis o rotura del músculo esquelético2. La rabdomiólisis puede estar causada por traumatismos severos, situaciones de isquemia prolongada, alteraciones metabólicas, actividad física intensa, abuso de alcohol y algunos compuestos tóxicos de origen químico o biológico3 (fig. 1). La incidencia de la rabdomiólisis no es del todo clara, pero se ha estimado que podría llegar al 7-10% de los pacientes que presentan un FRA3,4.
Hemoglobinuria
La hemoglobinuria es la presencia de Hb en orina como consecuencia de hemólisis intravascular. Este hecho provoca sobrecarga renal de Hb, sobre todo cuando la exposición a Hb libre es recurrente5. Entre las principales causas etiológicas de la hemoglobinuria destacan entidades hereditarias como la hemoglobinuria paroxística nocturna, la púrpura trombopénica trombocitopática, el síndrome hemolítico urémico (SHU), la anemia falciforme (AF), defectos en la membrana celular (eliptocitois, esferocitosis…), defectos enzimáticos (déficit de glucosa-6-fosfato deshidrogenasa, déficit de piruvato cinasa), anemias hemolíticas severas en el seno de reacciones transfusionales masivas, así como otras causas adquiridas de SHU y microangiopatías trombóticas de origen diverso6 (fig. 1).
Hemoproteínas y fracaso renal agudoEl FRA es una complicación frecuente en pacientes con hemoglobinuria o rabdomiólisis, especialmente si tienen enfermedad renal previa. Hasta un 50% de los pacientes con rabdomiólisis desarrolla FRA, dependiendo de la causa que la origine7,8. Así, la rabdomiólisis es una de las principales causas de FRA (5-25%) y llega a producir la muerte en un 2-46% de los casos, en ausencia de diálisis3,4. Situaciones asociadas a hemólisis intravascular también pueden inducir FRA en muchas ocasiones9,10.
Hemoproteínas y enfermedad renal crónicaLa aparición de enfermedad renal en pacientes con acumulación renal de hemoproteínas está bien documentada. Así, en enfermos con AF se ha descrito que la hemoglobinuria es un factor de riesgo independiente para la aparición y progresión de ERC11. Algo parecido ocurre en la hemoglobinuria paroxística nocturna, enfermedad en la que la ERC secundaria a la aparición de trombos en vasos renales y a la hemoglobinuria es una de sus complicaciones más importantes: puede afectar al 64% de los pacientes y causa el 18% de los fallecimientos12. En ausencia de tratamiento, el pronóstico del SHUa también es malo, con una mortalidad durante el brote del 25% y progresión hacia ERC en más de la mitad de los pacientes durante el año siguiente al diagnóstico13.
Mecanismos fisiopatológicos implicados en el daño renal por hemoproteínasEl principal efecto de las hemoproteínas en el riñón es su toxicidad directa sobre las células tubulares, independientemente de la causa que origine su liberación (hemo- o mioglobinuria) (fig. 2). En condiciones normales, la Hb se une a la haptoglobina y forman el complejo Hb-haptoglobina en el plasma14. Este complejo es demasiado grande para ser filtrado por el glomérulo y es degradado por el bazo, médula ósea e hígado. Sin embargo, en situaciones de hemólisis intravascular, la liberación masiva de Hb provoca que la haptoglobina se consuma. Como consecuencia, la Hb permanece más tiempo en el plasma y es más susceptible de disociarse en dímeros, los cuales son filtrados más fácilmente por el glomérulo. A diferencia de la Hb, la Mb atraviesa directamente la membrana de filtración glomerular debido a su menor tamaño molecular.

Una vez en la luz del túbulo, las hemoproteínas se pueden reabsorber por los túbulos proximales a través del complejo de receptores megalina/cubilina14 o bien degradarse liberando el grupo hemo y hierro libre, los cuales también tienen acciones deletéreas, tales como neutralización del óxido nítrico, vasoconstricción e isquemia15. El descenso en la biodisponibilidad del óxido nítrico provoca la desregulación de factores que controlan el tono vascular, como endotelina-1, tromboxano A2, factor de necrosis tumoral e isoprostanos16,17. La Hb y la Mb también son potentes agentes vasoconstrictores, ya que también reaccionan con el óxido nítrico, tal y como se ha descrito en enfermedades asociadas a hemólisis intravascular y rabdomiólisis18–20. En la luz del túbulo, tanto la Mb como la Hb son capaces de precipitar y formar agregados con la proteína Tamm-Horsfall y originar los cilindros hemáticos, los cuales provocan obstrucción intratubular en los segmentos distales de las nefronas21. Esta obstrucción se encuentra favorecida por el pH ácido de la orina, que incrementa la estabilidad de los enlaces entre las hemoproteínas y la proteína Tamm-Horsfall22,23.
En el interior de las células tubulares, las hemoproteínas se disocian liberando las globinas y el grupo hemo, el cual induce estrés oxidativo, muerte celular, así como la producción de citocinas inflamatorias y fibrosis, tal y como describiremos a continuación.
Estrés oxidativoLas hemoproteínas presentan distintas formas redox y son una fuente endógena de especies reactivas de oxígeno24. Cuando las hemoproteínas son captadas por las células tubulares, el grupo hemo es oxidado de Fe2+ a Fe3+ y da lugar a radicales hidroxilo25. En presencia de peróxidos, el Fe3+ se oxida a Fe4+ y genera radicales hidroperoxilo, los cuales son altamente reactivos y contribuyen a la formación de nuevas especies reactivas de oxígeno en el riñón26,27. Todos estos radicales promueven la peroxidación de lípidos de las membranas plasmáticas y generan malonaldehído, el cual interviene en la oxidación de proteínas y del material genético4,28,29. Este proceso da lugar a la producción de isoprostanos, citocinas proinflamatorias y a la expresión de moléculas de adhesión, lo que amplifica la respuesta inflamatoria30.
InflamaciónEl grupo hemo actúa como agonista de TLR-4 e induce una respuesta inflamatoria a través de la activación del factor de transcripción NF-kB31,32. A partir de su unión al pattern recognition receptor, la Hb promueve la activación de diversas señales de transducción como las cinasas c-Jun N-terminal, p38 y MAPK33. Otra de las vías implicadas está mediada por la activación del inflamasoma NLRP3 (nitrogen permease regulator like 3), responsable de la liberación de diferentes citocinas y quimiocinas implicadas en el reclutamiento de monocitos/macrófagos34. Así, se ha descrito la presencia de macrófagos de tipo proinflamatorio (M1) en fases tempranas en modelos experimentales de FRA por acumulación de hemoproteínas, que se diferencian a macrófagos antiinflamatorios (M2) en etapas posteriores35,36. Estos macrófagos M2 se encuentran presentes en biopsias renales de pacientes con rabdomiólisis, favismo, hemoglobinuria paroxística nocturna y brotes de hematuria macroscópica asociada a NIgA37–39.
Muerte celularSe han descrito distintos tipos de muerte celular en el epitelio tubular de pacientes y modelos experimentales asociados a acumulación de hemoproteínas4,39-43. La necrosis y apoptosis son los tipos de muerte que mejor han sido estudiados34,44–46. Los mecanismos moleculares que originan la muerte por apoptosis están asociados a una disfunción mitocondrial y a un aumento de proteínas proapotóticas (BAX y BAD), así como a la activación de la caspasa 3, la principal caspasa efectora34,47, y de proteínas de estrés de retículo endoplásmico48. Se han descrito otros tipos de muerte celular en estas enfermedades, tales como piroptosis (muerte celular mediada por la caspasa-1 que produce fragmentación del ADN y lisis celular) y ferroptosis (muerte celular dependiente de hierro). Así, se ha observado activación de caspasa-1 en modelos experimentales de rabdomiólisis34, mientras que el uso de inhibidores de ferroptosis en estos ratones redujo la muerte celular de túbulos proximales49. Por último, la acumulación de hemoproteínas y sus derivados pueden inducir autofagia como mecanismo de defensa46,50,51.
FibrosisLa fibrosis renal es otro de los mecanismos implicados en el daño renal por hemoproteínas. De hecho, pacientes con AF presentan fibrosis renal y un aumento en orina de TGF-β, uno de los principales mediadores profibróticos52. Aunque los fibroblastos y las células tubulares juegan un papel muy importante en la producción de proteínas de matriz extracelular, estudios recientes indican que los macrófagos podrían amplificar la respuesta profibrótica a partir de la producción de mediadores como CTGF y TGF-β en situaciones de rabdomiólisis35,36.
Los túbulos renales se consideran los principales sitios de toxicidad por Hb. Sin embargo, se ha descrito la presencia de proteinuria en modelos experimentales de exposición recurrente a hemoproteínas53, así como la presencia de glomeruloesclerosis focal y segmentaria en modelos experimentales de AF54 y en pacientes con hemólisis crónica y recurrente, tales como hemoglobinuria paroxística nocturna, SHU y AF55. Estos pacientes desarrollan proteinuria56 y padecen una disminución crónica de la filtración glomerular12,57. Estos datos señalan un vínculo entre la hemólisis intravascular y la disfunción glomerular. Sin embargo, los mecanismos fisiopatológicos no están claros. Se ha indicado que los cambios hemodinámicos asociados a esta enfermedad pueden ser responsables de la proteinuria y del daño renal progresivo, sin embargo, no hay pruebas definitivas de esta hipótesis58. Dado que la glomeruloesclerosis focal y segmentaria implica pérdida de podocitos, es posible que estas células también puedan sufrir daños mediados por hemoproteínas. En este sentido, datos sin publicar de nuestro grupo muestran que los podocitos son capaces de captar Hb, lo cual induce estrés oxidativo y muerte de estas células, así como pérdida de proteínas implicadas en el proceso de filtración glomerular, como sinaptopodina y nefrina.
Mecanismos de defensa frente a la toxicidad renal de las hemoproteínasExisten 2tipos de mecanismos de protección frente a los efectos nocivos de las hemoproteínas: los directos y los indirectos. Los mecanismos directos promueven el catabolismo de las hemoproteínas y derivados, mientras que los indirectos disminuyen el estrés oxidativo derivado de la presencia de estas moléculas, eliminando las especies reactivas de oxígeno o reparando los posibles daños ocasionados (fig. 3). A continuación, revisaremos cada uno de estos mecanismos de defensa en relación con el daño renal por hemoproteínas.

La haptoglobina (Hp) es una glicoproteína presente en el plasma en concentraciones elevadas (0,3-3g/L) y que es secretada principalmente por hepatocitos, aunque también se sintetiza en otros tejidos, incluido el riñón. La Hp se une a la Hb de forma irreversible e impide su filtración en el riñón59 y su translocación al endotelio60, lo que contrarresta sus efectos nocivos61. La Hp también puede unirse a Mb, aunque con menor afinidad que a la Hb62. La unión de Hb-Hp promueve la interacción y posterior internalización de este complejo a través del receptor de membrana CD163 presente en monocitos-macrófagos63. Los niveles de Hp están muy disminuidos en pacientes con hemólisis crónicas, tales como AF64, ya que la Hp se degrada tras ser endocitada65. Se ha descrito la importancia de esta proteína a partir de estudios en ratones knock out para Hp, los cuales son más sensibles al daño por hemólisis59. Estudios en modelos animales de AF o rabdomiólisis han demostrado que la administración de Hp reduce la vasooclusión31, el estrés oxidativo66 y el daño renal60,67,68.
CD163CD163 es un receptor presente en la superficie de monocitos circulantes y macrófagos, cuya función principal es el aclaramiento tisular de Hb63. CD163 tiene una alta afinidad por los complejos Hb-Hp, aunque también es capaz de unirse a la Hb libre69. Los macrófagos que expresan CD163 presentan una menor liberación de peróxido de hidrógeno e importantes funciones antiinflamatorias a través de la producción de IL-10 y estimulación de HO-170. Nuestro grupo ha observado un aumento de los macrófagos que expresan CD163 en biopsias renales de pacientes con hemólisis masivas, tales como hemoglobinuria paroxística nocturna 37 y favismo39. La expresión renal de CD163 estaba incrementada en áreas donde se acumulaba hierro y marcadores de estrés oxidativo. Recientemente, hemos descrito la presencia de CD163 en riñones de pacientes y modelos experimentales de rabdomiólisis35. Puesto que las funciones antiinflamatorias y antioxidantes de CD163 son bien conocidas, estos datos indican que CD163 puede ejercer un papel nefroprotector en respuesta a la acumulación renal de hemoproteínas.
HemoxigenasaLa hemoxigenasa (HO) es uno de los principales mecanismos protectores en situaciones de sobrecarga renal de Mb y Hb. La HO es la enzima responsable de degradar el grupo hemo, liberando biliverdina, Fe2+ y monóxido de carbono71, los cuales son potentes moléculas antiinflamatorias y antioxidantes que potencian los efectos beneficiosos de la HO72,73. Existen 3 isoformas de HO (HO-1, HO-2 y HO-3), que se diferencian en su distribución tisular, regulación y función. A diferencia de las otras isoformas, la expresión de HO-1 se induce en condiciones de estrés oxidativo y se expresa en muchos tejidos, incluido el riñón74. La expresión renal de HO-1 está aumentada en modelos experimentales de hemoglobinuria y rabdomiólisis, así como en pacientes con hemólisis intravascular66,75,76. El déficit de esta enzima en pacientes con hemólisis intravascular incrementa el daño tubular y glomerular77. En esta misma línea, animales knock out para HO-1 muestran que estos son más sensibles a la rabdomiólisis, presentan niveles más elevados de creatinina y mayor mortalidad78.
TransferrinaLa transferrina (Tf) es una glicoproteína secretada principalmente por el hígado y que se une al hierro libre para disminuir sus efectos adversos79. En función de la concentración de hierro, las células tubulares expresan el receptor de Tf (TfR1), que juega un papel esencial en el metabolismo de esta molécula a nivel renal80. Su expresión está regulada por las proteínas iron-regulatory proteins 1 y 2, altamente expresadas en los túbulos proximales, y que actúan como sensores de los niveles de hierro81–83. En condiciones normales, aproximadamente el 30% de los sitios de unión a hierro de la Tf se encuentran saturados, sin embargo, estos valores se incrementan cuando existen trastornos asociados a la acumulación de hierro84, como en el caso de hemocromatosis severa, en la que la saturación de la Tf excede el 60%85,86. Además, pacientes o modelos experimentales de hipotransferremia presentan niveles bajos de Tf, lo que promueve la sobrecarga renal de hierro87,88.
HemopexinaLa hemopexina (Hx) es una proteína plasmática que acompleja al grupo hemo para su posterior internalización y excreción hepática mediante su unión al receptor LRP1 (LDL receptor-related protein-1)89–91. En situaciones de hemólisis, la Hb se oxida y libera el grupo hemo al torrente circulatorio para posteriormente unirse a la albúmina sérica, que cede el grupo hemo a la Hx y libera el complejo en el hígado. Una vez en el hepatocito, el complejo Hx-hemo se degrada en el lisosoma, aunque una pequeña cantidad de Hx es reciclada y devuelta a la circulación. Por ello, en pacientes con eventos hemolíticos, la concentración plasmática de la Hx sérica disminuye92–95, mientras que se acumula en la corteza renal y aumentan sus niveles en orina96. La Hx ejerce un papel protector frente a los efectos nocivos del grupo hemo31,32,97. Así, ratones knock out para Hx presentan una peor recuperación de la función renal tras sufrir un evento de hemólisis intravascular, ya que presentan una mayor acumulación renal de hierro y, por tanto, mayores niveles de estrés oxidativo98,99.
FerritinaLa ferritina es una proteína compuesta por 24 subunidades que forman una estructura esférica hueca100. Tiene como principal función almacenar hierro en su interior, por lo que posee una capacidad protectora frente a la toxicidad causada por el hierro y las hemoproteínas. Tras la reacción catalizada por HO-1, el hierro se libera del grupo hemo y se almacena en el interior de la ferritina101. La expresión de ferritina está regulada por la concentración de hierro y la actividad de la HO-1102. Esta proteína juega un papel crucial en las enfermedades asociadas a hemoproteínas, ya que los ratones deficientes en ferritina muestran un daño renal notable103, y es un buen marcador sérico de AF104. Además, los niveles plasmáticos de Tf están aumentados en ratones deficientes para ferritina sometidos a rabdomiólisis103.
Nrf2Nrf2 es un factor de transcripción que controla la expresión de numerosos genes antioxidantes como HO-1 y ferritina105,106. En condiciones normales, Nrf2 se encuentra en el citoplasma unido a su represor Keap1, el cual es susceptible a cambios en el estado redox y está sujeto a degradación proteolítica a través del proteasoma. En presencia de estrés oxidativo, Nrf2 se libera de Keap1 y se transloca al núcleo, donde activa la expresión de genes antioxidantes107-112. La activación de Nrf2 resulta beneficiosa frente al daño renal asociado a acumulación de hemoproteínas en modelos experimentales y pacientes con anemia hemolítica113–115.
Mecanismos indirectosEste segundo grupo está formado por moléculas antioxidantes y diversas enzimas antioxidantes.
Mecanismos no enzimáticosExisten numerosas moléculas presentes en el organismo con actividad antioxidante, tales como vitaminas, melatonina o bilirrubina. Estas moléculas neutralizan los radicales libres y han sido implicadas en la protección frente al daño renal por hemoproteínas.
Un grupo de antioxidantes importante lo constituyen las vitaminas, entre ellas la vitamina C, que reacciona con el anión superóxido y peróxidos lipídicos, reduciendo el estrés oxidativo inducido por la lisis de eritrocitos in vitro116 e in vivo117. En esta línea, el tratamiento con vitamina C resultó efectivo en un contexto de FRA por hemoglobinemia en un paciente con deficiencia de glucosa-6 fosfato deshidrogenasa118. Los niveles de vitamina C disminuyen tras el desarrollo de rabdomiólisis y su administración redujo parcialmente las alteraciones histológicas así como la función renal en modelos experimentales de rabdomiólisis119. Otra vitamina importante es la vitamina E, ya que ejerce un papel relevante en el mantenimiento del balance redox y la integridad de las membranas celulares, actuando sobre los radicales peroxilo e hidroperoxilo. La administración de vitamina E inhibió la lisis de eritrocitos de pacientes con hemoglobinuria paroxística nocturna, lo que señala a esta vitamina como tratamiento efectivo para estos pacientes120,121. A diferencia de la vitamina C, el tratamiento con vitamina E no ha resultado ser tan efectivo en rabdomiólisis122.
La melatonina es una hormona secretada por la glándula pineal con numerosas propiedades antioxidantes ya que neutraliza radicales libres como el peróxido de hidrógeno, radical hidroxilo, peroxinitrito o el anión superóxido. Esta molécula también estimula la expresión de otras moléculas antioxidantes como superóxido dismutasa, glutatión peroxidasa y glutatión reductasa. Diversos estudios han mostrado un papel protector de esta hormona en modelos de FRA por rabdomiólisis o hemólisis intravascular, al disminuir la necrosis tubular y la peroxidación lipídica asociadas a ambas condiciones117,123.
El glutatión en su estado reducido (GSH) es un potente antioxidante celular que se puede oxidar a glutatión disulfuro (GSSG) por diversas reacciones enzimáticas. Diversos modelos experimentales de mio- o hemoglobinuria, así como estudios en pacientes con SHU y AF, muestran una reducción de los niveles de GSH a nivel renal117,123–128. La depleción de GSH incrementa la toxicidad mediada por estrés oxidativo en estas enfermedades, ya que el tratamiento con N-acetilcisteína, al restaurar los niveles de GSH, disminuye las alteraciones histológicas e inhibe la muerte celular asociada a estas condiciones129,130.
Mecanismos enzimáticosEste grupo encuadra moléculas con actividad enzimática que disminuyen el contenido de especies reactivas de oxígeno intracelular y, por tanto, protegen a la célula del daño oxidativo. La superóxido dismutasa (SOD) tiene la capacidad de dismutar el O2− en O2 y H2O2. Para la eliminación del H2O2, existen otras enzimas con actividad peroxidasa, como son la catalasa, la glutatión peroxidasa (GPx) y la tioxirredoxina reducida (TRX). La catalasa es una oxidorreductasa que cataliza la reacción de descomposición de H2O2 en O2 y agua. La Gpx cataliza la descomposición de H2O2 mediante la oxidación de GSH a GSSG y agua. La actividad de la GPx, catalasa y SOD se encuentra reducida en modelos experimentales de hemólisis intravascular117 y rabdomiólisis125,128,131–134. Además, los niveles plasmáticos de GPx y SOD se correlacionaron de forma negativa con la albuminuria en pacientes con AF135. Finalmente, la TRX protege del daño renal asociado a rabdomiólisis mediante la reducción del estrés oxidativo y la inflamación136.
TratamientosEn la actualidad no existe ningún tratamiento específico dirigido a evitar el daño inducido por la ferroproteínas en sus diferentes formas de presentación clínica. La alcalinización de la orina podría resultar beneficiosa al disminuir la disociación del hierro presente en las hemoproteínas. Esta alcalinización se puede realizar con bicarbonato oral con monitorización del pH urinario y sérico. Sin embargo, un claro beneficio no ha sido fehacientemente demostrado. El empleo de bloqueantes de los canales del calcio en modelos experimentales ha demostrado un incremento en la excreción urinaria de hierro por mecanismos aún desconocidos, lo que provoca, en última instancia, una disminución de la acumulación de hierro a nivel renal137.
El uso de los quelantes de hierro en enfermedades asociadas a acumulación de esta molécula reduce el daño oxidativo138, además de evitar el depósito de hierro139. La desferrioxamina administrada profilácticamente disminuye el estrés oxidativo derivado de la presencia de Hb140. Los quelantes de hierro disminuyen la toxicidad producida por el depósito masivo de hierro en pacientes politransfundidos141, aunque trabajos recientes cuestionan su capacidad nefroprotectora en el FRA inducido por hemoglobina142. Debemos recordar que ciertos quelantes del hierro, como el deferasirox143 o la deferroxamina144, son potenciales agentes nefrotóxicos, lo que obliga a realizar un seguimiento estrecho durante su uso145.
El tratamiento profiláctico con antioxidantes como la N-acetilcisteína ha dado buenos resultados en la prevención del daño tubular secundario a mio- o hemoglobinuria146. Otros antioxidantes, como el acetaminofen, también resultan efectivos27,147. El posible papel protector de la vitamina E148 o C27, así como polifenoles149, flavonoides128,150–152 o la L-carnitina153 también ha sido investigado, con resultados dispares, tal y como comentamos anteriormente. Recientes estudios también han abordado el uso de células madre, con resultados positivos, sobre todo en modelos de rabdomiólisis154.
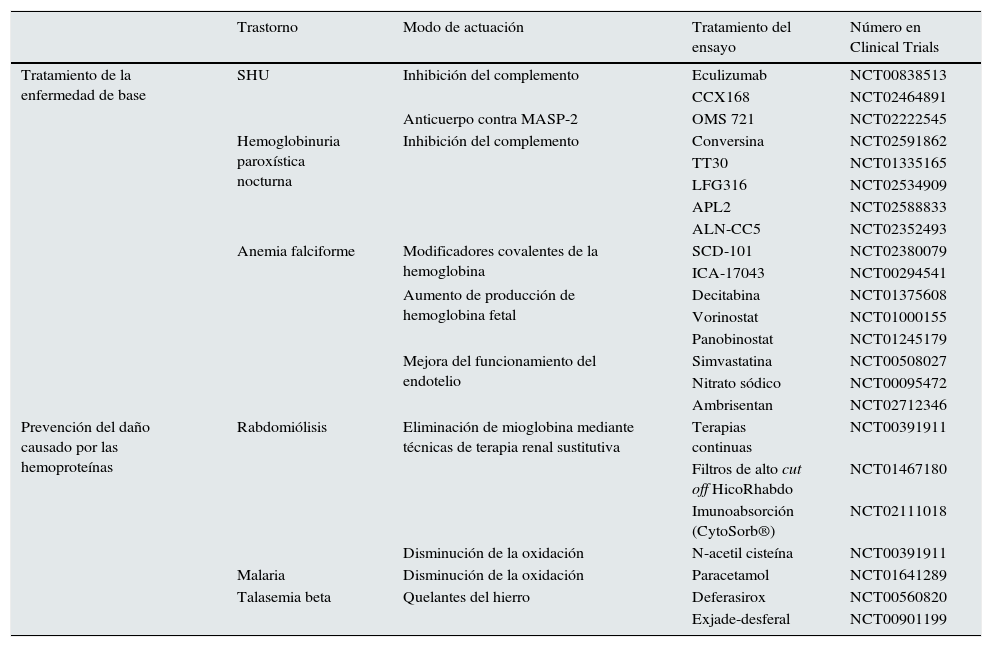
Ensayos clínicosEn el momento actual los ensayos clínicos en marcha para el tratamiento de los trastornos ocasionados por hemoproteínas se centran en 2aspectos. El primero consiste en tratar la enfermedad de base para evitar la liberación a plasma de las hemoproteínas y el segundo en paliar el daño que las hemoproteínas pueden ocasionar una vez liberadas. De este modo, en enfermedades como el SHU atípico y la hemoglobinuria paroxística nocturna, la mayoría de los ensayos prueban fármacos que actúan sobre el sistema del complemento, principalmente eculizumab, o bien nuevas moléculas que actúan a otros niveles. Entre ellas destacan: CCX168 (antagonista de C5aR); conversina (proteína que evita la acción sobre C5 de su convertasa), TT30 (ALXN1102 y ALXN1103; proteínas recombinantes que contienen los dominios 1-5 del factor H y disminuyen la actividad de las convertasas del complemento y activan el factor I), LFG316 (anticuerpo monoclonal anti-C5), APL2 (inhibidor de C3) y ALN-CC5 (inhibidor a nivel hepático de la síntesis de C5) (tabla 1). Además, en el SHUa se están desarrollando anticuerpos contra MASP-2 llamados OMS 723. En AF existen ensayos con fármacos como SCD 101 o ICA-17043 (evita que los hematíes se conviertan en falciformes), con decitabina, vorinostat y panobinostat (para aumentar la hemoglobina fetal) y con estatinas, nitrato sódico y ambrisentan (antagonista selectivo del receptor de endotelina A) para mejorar la disfunción endotelial, mantener una buena perfusión tisular en las crisis y evitar la rotura de los hematíes. SCD 101 también se ha usado en la talasemia beta.
Ensayos clínicos en enfermedades asociadas a acumulación renal de hemoproteínas
| Trastorno | Modo de actuación | Tratamiento del ensayo | Número en Clinical Trials | |
|---|---|---|---|---|
| Tratamiento de la enfermedad de base | SHU | Inhibición del complemento | Eculizumab | NCT00838513 |
| CCX168 | NCT02464891 | |||
| Anticuerpo contra MASP-2 | OMS 721 | NCT02222545 | ||
| Hemoglobinuria paroxística nocturna | Inhibición del complemento | Conversina | NCT02591862 | |
| TT30 | NCT01335165 | |||
| LFG316 | NCT02534909 | |||
| APL2 | NCT02588833 | |||
| ALN-CC5 | NCT02352493 | |||
| Anemia falciforme | Modificadores covalentes de la hemoglobina | SCD-101 | NCT02380079 | |
| ICA-17043 | NCT00294541 | |||
| Aumento de producción de hemoglobina fetal | Decitabina | NCT01375608 | ||
| Vorinostat | NCT01000155 | |||
| Panobinostat | NCT01245179 | |||
| Mejora del funcionamiento del endotelio | Simvastatina | NCT00508027 | ||
| Nitrato sódico | NCT00095472 | |||
| Ambrisentan | NCT02712346 | |||
| Prevención del daño causado por las hemoproteínas | Rabdomiólisis | Eliminación de mioglobina mediante técnicas de terapia renal sustitutiva | Terapias continuas | NCT00391911 |
| Filtros de alto cut off HicoRhabdo | NCT01467180 | |||
| Imunoabsorción (CytoSorb®) | NCT02111018 | |||
| Disminución de la oxidación | N-acetil cisteína | NCT00391911 | ||
| Malaria | Disminución de la oxidación | Paracetamol | NCT01641289 | |
| Talasemia beta | Quelantes del hierro | Deferasirox | NCT00560820 | |
| Exjade-desferal | NCT00901199 |
Por otro lado, una vez que las hemoproteínas se han liberado al plasma, los ensayos se centran en 2estrategias. La primera consiste en intentar retirar dichas moléculas del plasma. Así se ha hecho en algunos ensayos en pacientes con rabdomiólisis que precisan de terapia renal sustitutiva en los que se han analizado los efectos de técnicas continuas, hemofiltros de alto cut off o técnicas de inmunoabsorción (CytoSorb®) para eliminar lo más rápidamente posible la Mb del plasma. La segunda estrategia se basa en disminuir su efecto tóxico. Para ello se está intentando evitar la oxidación inducida por hemoglobinuria en la malaria con paracetamol y la inducida por mioglobinuria con N-acetilcisteína. También hay ensayos en marcha que emplean quelantes de hierro (deferasirox o la combinación exjade-desferal) en la talasemia para evitar el depósito de este elemento en los órganos diana.
ConclusiónLa acumulación de hemoproteínas en el riñón resulta nefrotóxica. Actualmente disponemos de evidencias que muestran efectos adversos no solo a corto plazo, sino pérdida crónica de la función renal. Aunque se han descrito numerosos efectos adversos de estas moléculas, es necesario seguir caracterizando los mecanismos patogénicos de las hemoproteínas para identificar nuevas dianas terapéuticas y evitar sus efectos adversos. En este sentido, los podocitos podrían ser nuevas dianas celulares de los efectos nocivos de las hemoproteínas. Desde el punto de vista terapéutico, los datos de los que disponemos en la actualidad se basan fundamentalmente en estudios en modelos animales. Medidas terapéuticas dirigidas a disminuir la mio- o la hemoglobinuria serían de gran importancia para evitar el daño renal por estas moléculas.
FinanciaciónEste trabajo ha sido financiado por una ayuda de la Sociedad Española de Nefrología a Investigación en Nefrología, Fondo de Investigaciones Sanitarias (FIS) (ISCIII/FEDER) (Programa Miguel Servet: CP10/00479 y CPII16/00017; PI13/00802 and PI14/00883) y Fundación Renal Íñigo Álvarez de Toledo (FRIAT) a JAM, Fundación Conchita Rábago a MGH, REDinREN (RD012/0021), FIS PI13/02502 y ICI14/00350 a MP, y FIS/FEDER PI14/00386 y Centro de Investigación Biomédica en Red de Diabetes y Enfermedades Metabólicas asociadas (CIBERDEM) a JE.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



