
The activation of the alternative pathway of the complement is involved in the development of several renal diseases, such as atypical haemolytic uraemic syndrome and C3 glomerulopathy. In C3 glomerulopathy, a high percentage of patients have circulating levels of the autoantibody called C3NeF, which causes systemic dysregulation of the complement system. In some cases, the presence of this antibody has been related with abnormalities of adipose tissue, causing acquired partial lipodystrophy (Barraquer–Simons syndrome). Acquired partial lipodystrophy is an extremely rare disorder affecting the distribution of subcutaneous adipose tissue and that mainly onsets during childhood. These patients, in addition to possibly presenting with all the metabolic disorders associated with the adipose tissue defect, present with C3 hypocomplementemia and C3NeF and 25% have developed C3 glomerulopathy. Although it has been known for some time how the dysregulation of the complement system affects the kidneys, it remains unknown how it exactly affects adipose tissue; nevertheless, the relationship is quite clear. In this paper, we describe the connection between the complement system with the biology of the adipose tissue and its pathogenesis reflected from acquired partial lipodystrophy.
La activación de la vía alternativa del complemento interviene en el desarrollo de varias enfermedades renales, como el síndrome hemolítico urémico atípico o la glomerulopatía C3. En esta última enfermedad un elevado porcentaje de los pacientes presentan niveles circulantes de un autoanticuerpo denominado C3NeF, causante de la desregulación del complemento a nivel sistémico. En ciertos casos, la presencia de este anticuerpo se asocia con alteraciones en el tejido adiposo, causando lipodistrofia parcial adquirida (síndrome de Barraquer-Simons), una enfermedad ultra-rara que afecta a la distribución del tejido adiposo subcutáneo y que comienza principalmente durante la infancia. Estos pacientes, además de poder presentar los problemas metabólicos asociados al defecto en el tejido adiposo, presentan hipocomplementemia C3 junto con la presencia de C3NeF y, en un 25% de los casos desarrollan una glomerulopatía C3. Aunque se sabe desde hace tiempo cómo la desregulación del sistema del complemento afecta al riñón, se desconoce de forma precisa cómo lo hace en el tejido adiposo; no obstante, su relación está bastante clara. En este artículo se va a describir la relación del sistema del complemento con la biología del tejido adiposo y su patogenia reflejada a partir de la lipodistrofia parcial adquirida.
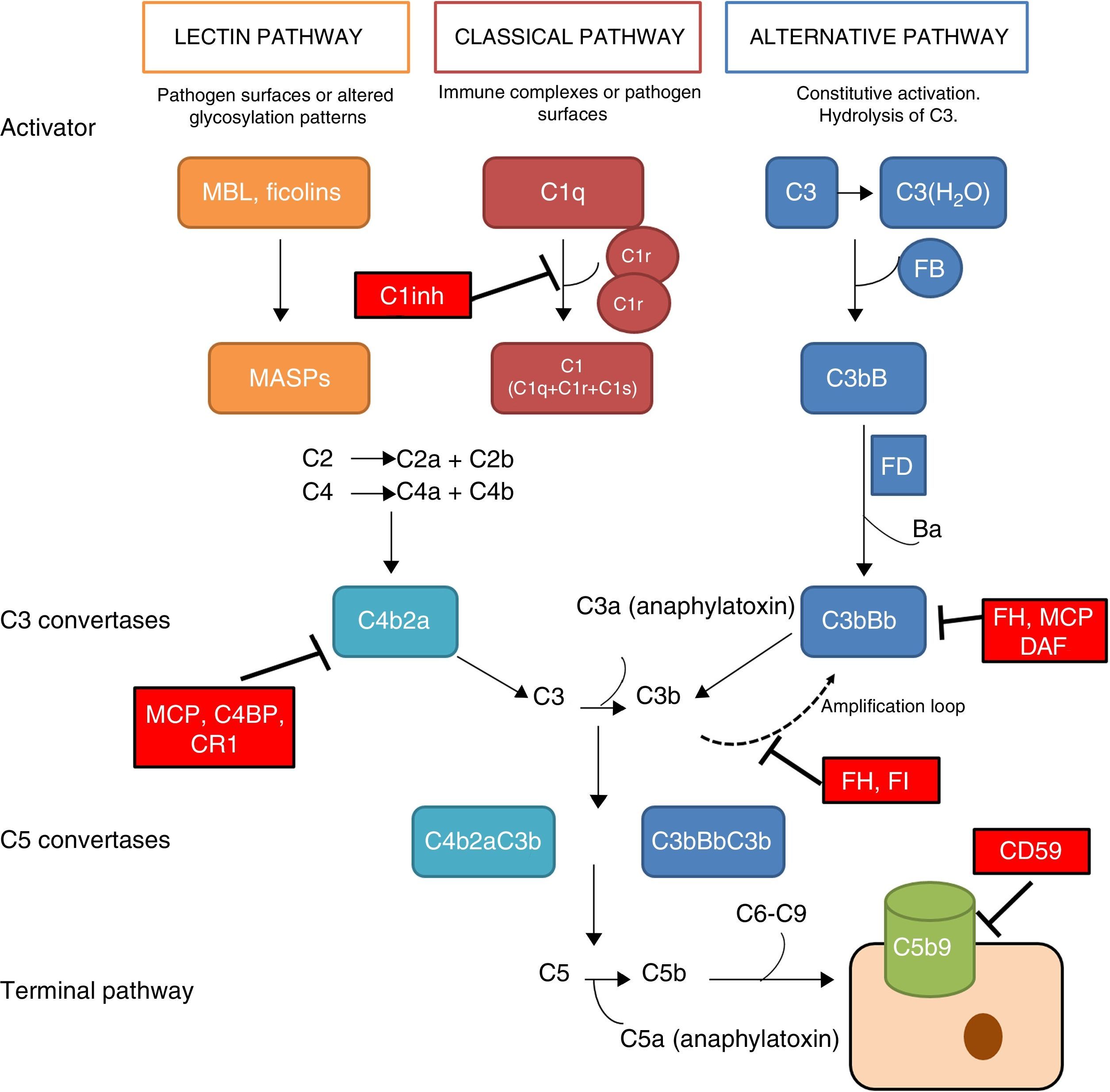
The complement system is a fundamental component of innate immunity playing a crucial role in the defence against infections, elimination of apoptotic cells, processing of immune complexes and modulation of adaptive immunity. The complement is a complex molecular system capable of triggering warning signals in the presence of foreign agents, differentiating between the body's own components and foreign components. Through a molecular tagging system, it is capable of identifying these foreign components to be eliminated by opsonophagocytosis or to destroy them via direct cell lysis. Complement activation takes place through three different activation pathways: the classical pathway, the alternative pathway and the lectin pathway (Fig. 1). These three pathways, closely related phylogenetically, differ in their mechanisms of activation and in the initial activation steps, but all of them converge in the formation of multi-molecular enzymatic complexes responsible for the activation of the C3 component, the C3 convertase.1
 that cleaves C3 into C3a and C3b. The C3b generated by any convertase can, in turn, form more alternative pathway convertase, through which amplification of complement activation occurs. The binding of a new molecule of C3b to the C3 convertases confers the ability to cleave C5 into C5a and C5b. C5b initiates the terminal complement pathway, which eventually leads to the formation of the membrane attack complex (C5b-9) and lysis of the target cells. Complement activation is controlled at various levels by different soluble and membrane regulatory proteins.")
The complement system. The complement system can be activated by three pathways. Activation by any of the three pathways leads to the generation of C3 convertase (C4b2b or C3bBb) that cleaves C3 into C3a and C3b. The C3b generated by any convertase can, in turn, form more alternative pathway convertase, through which amplification of complement activation occurs. The binding of a new molecule of C3b to the C3 convertases confers the ability to cleave C5 into C5a and C5b. C5b initiates the terminal complement pathway, which eventually leads to the formation of the membrane attack complex (C5b-9) and lysis of the target cells. Complement activation is controlled at various levels by different soluble and membrane regulatory proteins.
The classical pathway is activated mainly by the binding of C1q to antigen–antibody complexes, and the activation of the lectin pathway mainly occurs through to the recognition of mannose groups, which are characteristic of bacterial surfaces. Activation of these two pathways results in the generation of a protein complex with enzymatic activity: the C3 convertase of the classical/lectin pathways (C4b2b) capable of activating the C3 molecule, cleaving it into C3a and C3b.
The alternative pathway is constitutively active, due to the spontaneous activation of C3 in plasma through the ‘tick-over’ mechanism. The activation of C3 generates C3a and C3b fragments; the C3b can be associated with factor B (FB) to form C3 convertase of the alternative pathway (C3bB) in an inactive state. Each C3bB complex is activated through the proteolytic cleavage of FB by factor D (FD), resulting in fragments Ba and Bb. The resulting complex, C3bBb, is capable of amplifying the system by means of positive feedback to generate thousands of C3b molecules in a very short time.1 The C3bBb convertase is unstable and needs to be stabilised by means of the positive regulator, properdin, which increases the half-life of this multi-molecular complex and serves as a focal point for the local amplification of the complement system.2 The incorporation of C3b molecule to a C3 convertase results in the formation of C5 convertase. This is capable of cleaving C5, generating C5a, a potent anaphylatoxin, and C5b, which initiate in the cell surface, along with C6, C7, C8 and C9, the generation of the membrane attack complex (C5b9).
The rapid and effective dissociation of the C3bBb complex and the inactivation of C3b is a critical step for the homeostasis of the complement system, and to prevent tissue damage when it is activated. These functions are carried out by a series of soluble regulatory proteins (factor H and factor I) and of membrane regulatory proteins (MCP, DAF, CR1 and CD59) (Fig. 1). The importance of the integrity of this system reveals why the deficiencies of some of its components may lead to situations that can be fatal. Problems also arise due to an inefficient regulation, which may cause indiscriminate activation and generation of circulating fragments or tissue injury.3
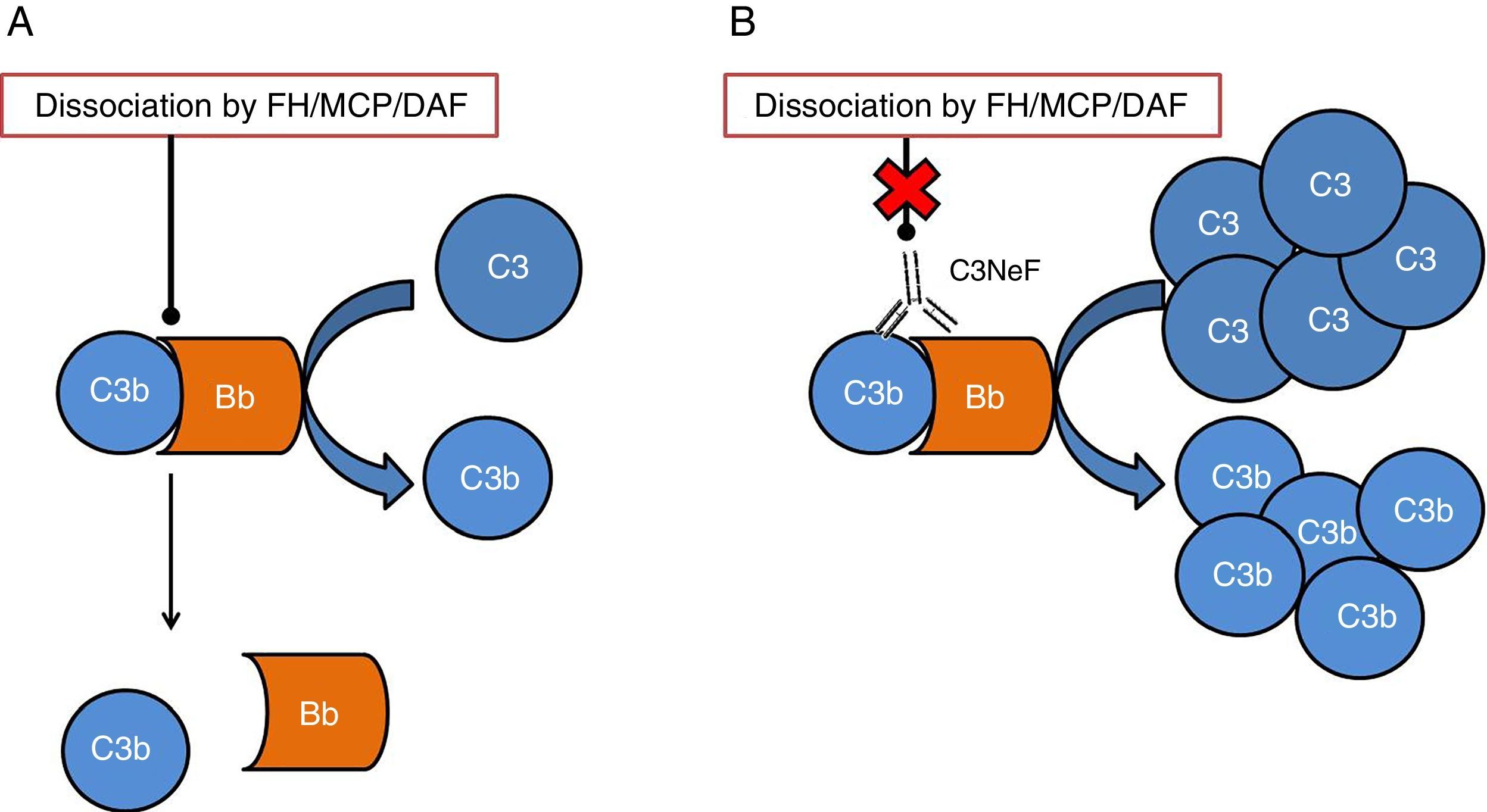
One of the tissues which is most affected by dysregulation of the complement system is the kidney. The surfaces of the glomerulus are very sensitive to the inflammatory effects caused by complement activation both at a systemic and local level. The two conditions which have most commonly been linked to alterations of the complement system are C3 glomerulopathy (C3G) and atypical haemolytic uraemic syndrome (aHUS). In both cases, mutations have been found in genes of components or regulators of the alternative pathway, thus the activation of this pathway cannot be controlled and the tissue is affected.4 In addition to the mutations, there are autoantibodies that are directly related to the onset of these diseases. These autoantibodies have a negative effect on the correct regulation of the alternative pathway.5 One of the most well-known autoantibodies is C3 nephritic factor (C3NeF), an antibody which is capable of binding with high affinity to C3 convertase of the alternative pathway (C3bBb), making it significantly more stable increasing its half-life (Fig. 2). This antibody is more common in the C3G, being found in 80% of patients with dense deposit disease and in 40–50% of patients with C3 glomerulonephritis.6
 In physiological conditions, C3 convertase cleaves the C3 molecule into C3a and C3b, in a process regulated by proteins such as factor H (FH), MCP or DAF. (B) When C3NeF binds to the convertase, it prevents the FH, MCP or DAF regulators from dissociating the complex, remaining active for longer and causing an increase in consumption of C3.")
Diagram of the effect of C3NeF on the C3 convertase of the alternative pathway. (A) In physiological conditions, C3 convertase cleaves the C3 molecule into C3a and C3b, in a process regulated by proteins such as factor H (FH), MCP or DAF. (B) When C3NeF binds to the convertase, it prevents the FH, MCP or DAF regulators from dissociating the complex, remaining active for longer and causing an increase in consumption of C3.
Another pathological situation where C3NeF is highly prevalent is in acquired partial lipodystrophy (also known as Barraquer–Simons syndrome), an extremely rare disorder of the subcutaneous adipose tissue, in which 25% of patients develop C3G at medium-to-long term.7 This article will review the role of the complement system in the biology of adipose tissue and in the pathogenesis of acquired partial lipodystrophy, as well as its relationship with C3G. Lastly, existing therapeutic regimens, including anti-complement therapy, will be discussed.
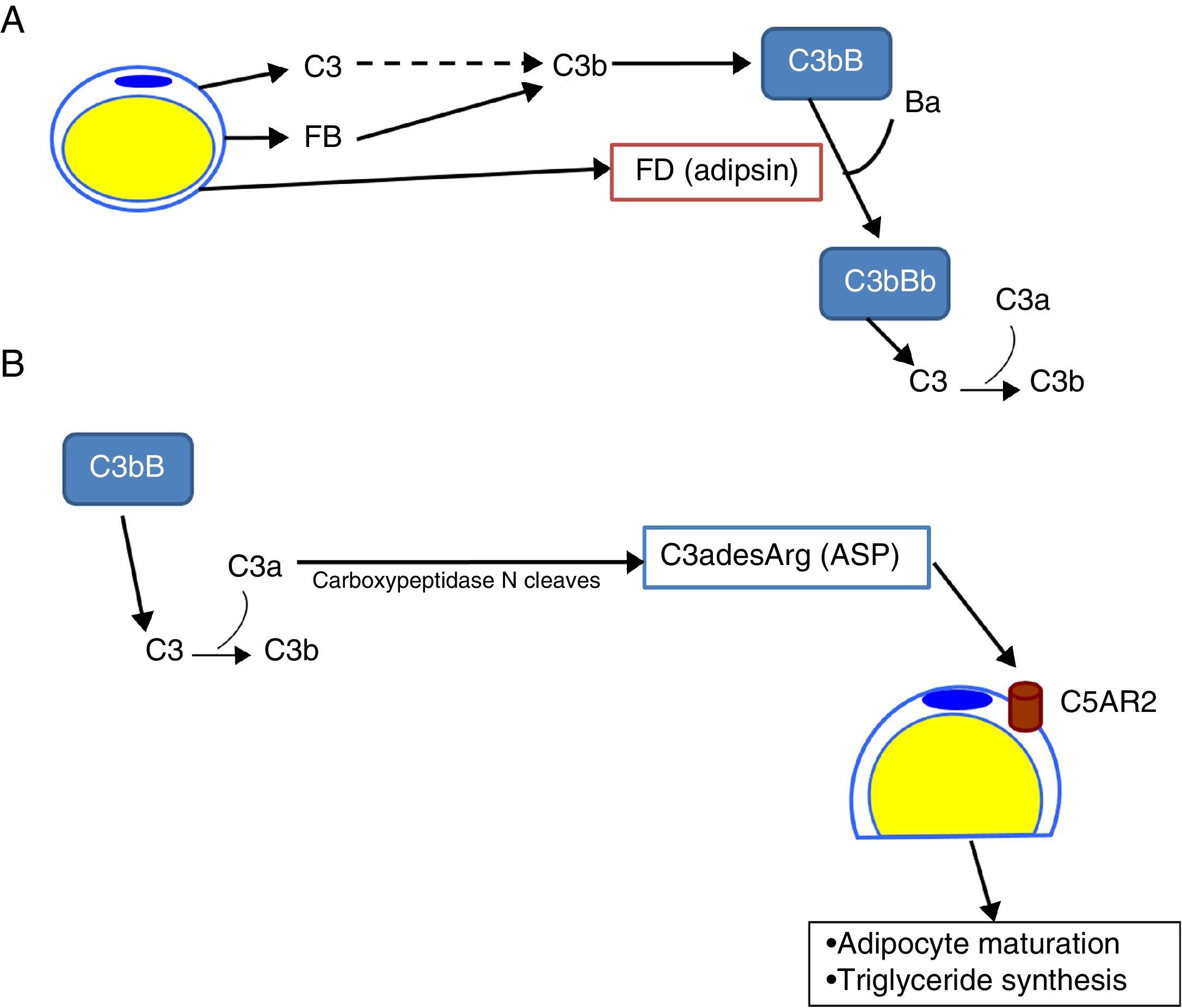
The complement system in the biology of the adipocyteDr Spiegelman's group demonstrated that murine adipocytes synthesized and secreted a protein that they called adipsin, which has some relevant effect on adipocyte development.8,9 Later on, a protein with a homology greater than 98% with adipsin was found in humans. This protein was identified as FD. It was already known that FD had some key functions in the activation of the alternative complement pathway.10 In addition, unlike most of the complement proteins, which are synthesized in the liver, 95% of circulating FD is synthesized in adipose tissue.11 Adipocytes also synthesise C3, FB12 and properdin.13 These data show the importance of the alternative pathway in adipose tissue, with adipocytes being capable of generating C3 convertase (C3bBb) on their surface, enriching the extracellular space with C3a, a potent proinflammatory anaphylatoxin (Fig. 3A). In an independent study, Sniderman and Cianflone described the presence of a factor in serum capable of stimulating the synthesis of triglycerides in human adipose tissue. This was named acylation stimulating protein (ASP). After sequencing it, they discovered that ASP corresponded to C3desArg,14 a degradation product of C3a by carboxypeptidase N. Subsequent studies revealed that the complement receptor, C5AR2, was present on the surface of adipocyte cell membranes, which binds ASP to stimulate the synthesis of triglycerides and promote the maturation of preadipocytes in in vitro models15,16 (Fig. 3B).
![Role of the complement system in the biology of the adipocyte. (A) Adipocytes secrete complement components, such as C3, factor B (FB) and factor D (FD, adipsin) and are capable of generating a C3 convertase (C3bBb) of the alternative pathway in their surroundings. (B) C3a is cleaved by activation of the C3 molecule. In turn, C3a is converted into C3adesArg (acylation stimulating protein [ASP]) due to the action of adipose tissue carboxypeptidase N. C3adesArg acts as a ligand for its receptor, C5AR2, which is located on the surface of adipocytes, the main function of this molecule is to signal the stimulation of triglyceride synthesis during adipose tissue maturation.](https://static.elsevier.es/multimedia/20132514/0000003800000003/v1_201805170414/S2013251418300609/v1_201805170414/en/main.assets/gr3.jpeg?xkr=ue/ImdikoIMrsJoerZ+w92lL8PRnKGMnsiVXV6EVJ5QRkjJZIKj2umwUyY+cL6K8cyMCoG0zLrx/ORKa7YTjKbqFEVsSFQNrNrBVPYmQ84IisQ5BuRZU6fCU5dHtJRHjriW8EhMDfpMZ4HE/MaHWTLJ3s/DWkheOs4Xbr8KhShPRAKETxZ1OHzXbd7HmevlFx83rxjxE1qVQe5716Af0ltYcQF+ur4BXA6B9b6WysnFrl4dOJ2vSAllxQV+7UNKUMuTLqBG1JNkLhGgBgc96ofYWkOQuo0yRwSNwWCKHnxMZP3deoPUsDrbHWpNGu/ri "Role of the complement system in the biology of the adipocyte. (A) Adipocytes secrete complement components, such as C3, factor B (FB) and factor D (FD, adipsin) and are capable of generating a C3 convertase (C3bBb) of the alternative pathway in their surroundings. (B) C3a is cleaved by activation of the C3 molecule. In turn, C3a is converted into C3adesArg (acylation stimulating protein [ASP]) due to the action of adipose tissue carboxypeptidase N. C3adesArg acts as a ligand for its receptor, C5AR2, which is located on the surface of adipocytes, the main function of this molecule is to signal the stimulation of triglyceride synthesis during adipose tissue maturation.")
Role of the complement system in the biology of the adipocyte. (A) Adipocytes secrete complement components, such as C3, factor B (FB) and factor D (FD, adipsin) and are capable of generating a C3 convertase (C3bBb) of the alternative pathway in their surroundings. (B) C3a is cleaved by activation of the C3 molecule. In turn, C3a is converted into C3adesArg (acylation stimulating protein [ASP]) due to the action of adipose tissue carboxypeptidase N. C3adesArg acts as a ligand for its receptor, C5AR2, which is located on the surface of adipocytes, the main function of this molecule is to signal the stimulation of triglyceride synthesis during adipose tissue maturation.
Lipodystrophies are a heterogeneous set of very rare conditions characterised by deficiency of adipose tissue in the absence of nutritional deprivation or catabolic state.7 Loss of adipose tissue may be “generalized” affecting the entire body, it may be “partial” affecting only well-defined regions, or “localized” to small areas under the skin. More than 1000 cases have been reported worldwide, with a prevalence of less than 1:1,000,000, although this is likely to be underestimated. The above-mentioned types may separated according to their aetiology: genetic (familial) or acquired lipodystrophies.17 Unlike localized lipodystrophies, the partial or generalized forms mean that patients are predisposed to develop resistance to insulin and to complications associated with this status, such as diabetes mellitus, hypertriglyceridaemia, hepatic steatosis, polycystic ovary syndrome and acanthosis nigricans. The severity of these complications will depend on the subtype, the extent of fat loss, age and gender.18
The main subtypes of lipodystrophies include: congenital generalized lipodystrophy; familial partial lipodystrophy; acquired generalized lipodystrophy and acquired partial lipodystrophy. There are other systemic diseases which are associated with lipodystrophy, such as progeroid syndromes and autoinflammatory diseases.18
Although genetic testing is not necessary for diagnosis, it is very useful to identify subtypes of familial lipodystrophies. Genetic tests can help to identify family members at risk, especially for subtypes of lipodystrophy associated with subtle physical phenotypes or those with a high risk of morbidity and mortality (for example, cardiomyopathy and arrhythmias). A negative result does not necessarily mean that the individual does not have the disease, as there are forms of lipodystrophies for which the genetic basis is unknown.
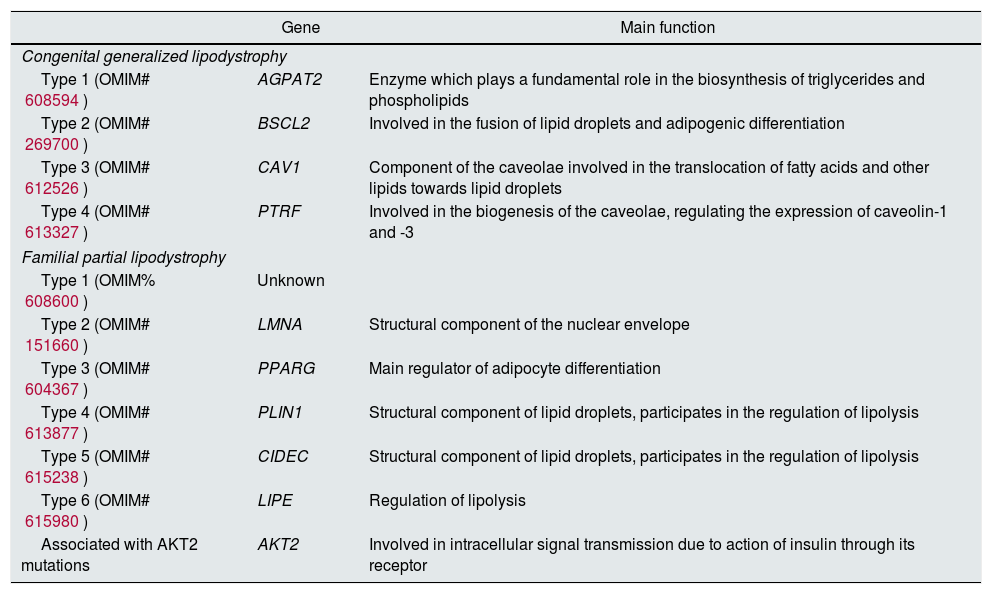
Most genes mutations reported encode for proteins involved in the differentiation/survival of the adipocyte, or in the process of the formation of lipid droplets. Congenital generalized lipodystrophy is caused by autosomal recessive mutations in AGPAT2, BSCL2, CAV1 and PTRF. By contrast, familial partial lipodystrophy is caused mainly by autosomal dominant mutations in LMNA, PPARg, PLIN1 and AKT2, although there are two types with recessive mutations in CIDEC and LIPE.17Table 1 refers to all the subtypes of existing congenital lipodystrophies, the associated gene and its main function.
Main congenital lipodystrophies: classification, causal genes and main function.
| Gene | Main function | |
|---|---|---|
| Congenital generalized lipodystrophy | ||
| Type 1 (OMIM# 608594) | AGPAT2 | Enzyme which plays a fundamental role in the biosynthesis of triglycerides and phospholipids |
| Type 2 (OMIM# 269700) | BSCL2 | Involved in the fusion of lipid droplets and adipogenic differentiation |
| Type 3 (OMIM# 612526) | CAV1 | Component of the caveolae involved in the translocation of fatty acids and other lipids towards lipid droplets |
| Type 4 (OMIM# 613327) | PTRF | Involved in the biogenesis of the caveolae, regulating the expression of caveolin-1 and -3 |
| Familial partial lipodystrophy | ||
| Type 1 (OMIM% 608600) | Unknown | |
| Type 2 (OMIM# 151660) | LMNA | Structural component of the nuclear envelope |
| Type 3 (OMIM# 604367) | PPARG | Main regulator of adipocyte differentiation |
| Type 4 (OMIM# 613877) | PLIN1 | Structural component of lipid droplets, participates in the regulation of lipolysis |
| Type 5 (OMIM# 615238) | CIDEC | Structural component of lipid droplets, participates in the regulation of lipolysis |
| Type 6 (OMIM# 615980) | LIPE | Regulation of lipolysis |
| Associated with AKT2 mutations | AKT2 | Involved in intracellular signal transmission due to action of insulin through its receptor |
AGPAT2: 1-acylglycerol-3-phosphate O-acyltransferase 2; AKT2: v-akt murine thymoma viral oncogene homolog 2; BSCL2: Berardinelli-Seip congenital lipodystrophy 2; CAV1: caveolin-1; CIDEC: cell death-inducing DFFA-like effector c; LIPE: hormone sensitive lipase; LMNA: lamin A/C; PLIN1: perilipin; PPARG: peroxisome proliferator-activated receptor gamma; PTRF: polymerase I and transcript release factor.
Acquired lipodystrophies are more common than the familial type. Acquired partial lipodystrophy (OMIM# 613913) was the first to be described more than 130 years ago.19 This syndrome is characterised by the absence of subcutaneous adipose tissue in the face, upper extremities and trunk, with hypertrophy of fat on the lower extremities. Females are affected up to four times more than males. The mean age of onset of this lipodystrophy is seven years. There is no uniformity regarding the reported prevalence of metabolic disorders, but it is clear is that acquired partial lipodystrophy is associated with these disorders far less frequently than other familial partial lipodystrophies (43%), and differs greatly from all the generalized types (50–80%).20 In this type of lipodystrophy, 75–90% of patients present very low levels of complement component C3 and this is closely related to detectable titres of C3NeF18 in more than 80% of patients. In this regard, given that the presence of this autoantibody is strongly associated with the development of C3G, it is reasonable that a quarter of patients with this lipodystrophy end up developing glomerulopathy. The relationship between kidney disease and partial lipodystrophy was described by Gellis et al. (1958),21 and, subsequently, it was associated with mesangiocapillary glomerulonephritis by other authors.22 Within the spectrum of C3G, the most common form of presentation is dense deposit disease,20 although there are some cases in which the histological study has revealed IgA nephropathy.23 It is interesting, particularly for adequate clinical control of these patients, to have in mind that the time interval from the onset of lipodystrophy to the development of nephropathy is approximately eight years.20 In other series, this period is increased to up to 10 years, and some cases are even reported 20 years.22 It is therefore advisable to monitor patients to prevent or act promptly against the nephropathy.
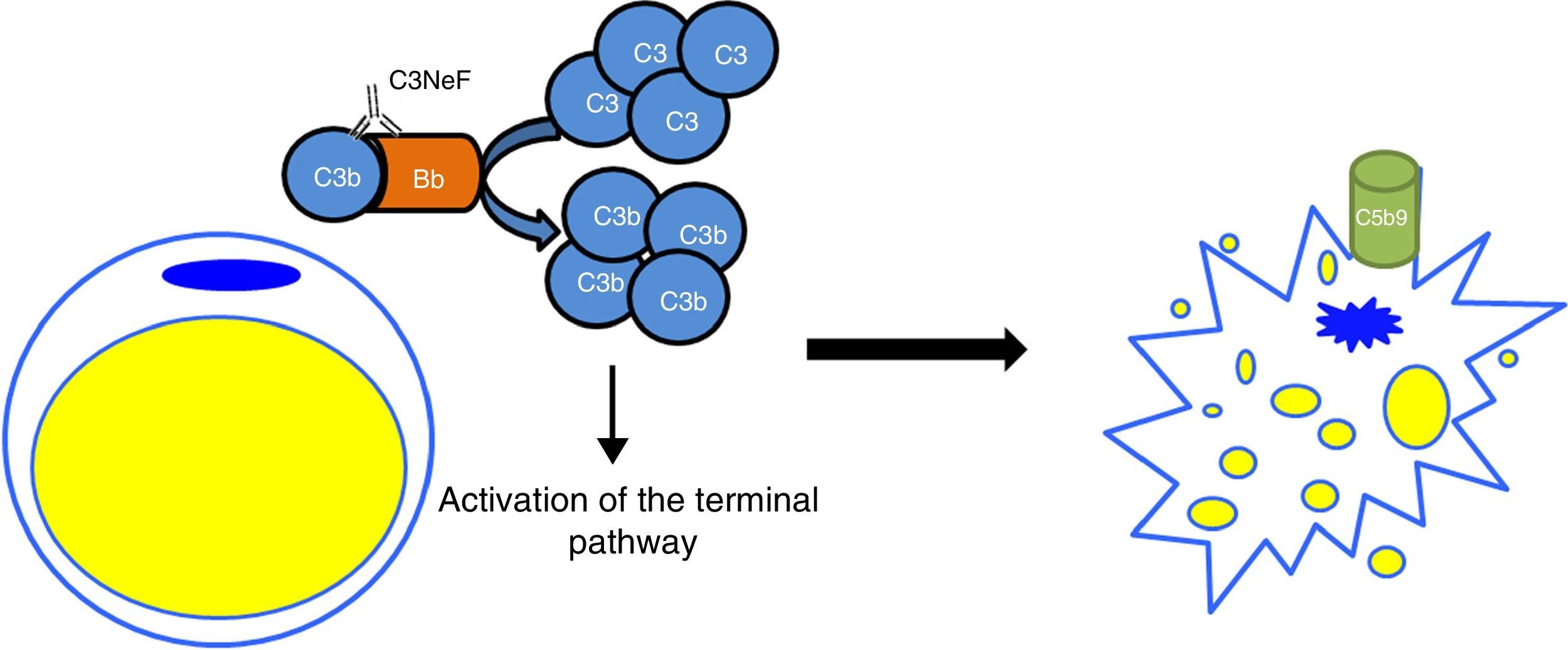
In the C3G cohorts, only a low percentage of patients would also present with acquired partial lipodystrophy. However, among patients with lipodystrophy, the frequency of finding also C3G is high. In a study of 125 patients with membranoproliferative glomerulonephritis, 11 had C3 hypocomplementaemia, but only two were diagnosed with lipodystrophy.24 The only link between both conditions is C3 hypocomplementaemia and the presence of C3NeF. It was hypothesised that a dysregulation of the alternative pathway as a consequence of the presence of C3NeF could result in a fat disorder, due mainly to complement-dependent cytotoxicity. This possible link was analysed by Mathieson and his team, who proved that the serum of a patient with acquired partial lipodystrophy, positive for C3NeF, was capable of causing lysis of adipocytes, while the serum of a healthy donor was not capable of doing so.25 Therefore, the currently accepted pathogenic mechanism is that C3NeF is capable of utilising the capacity of the adipocyte to form C3 convertase in its surroundings and inducing complement-mediated lysis (Fig. 4). Despite this, the pathophysiological mechanism of C3NeF in acquired partial lipodystrophy is unknown and, to some extent, debatable. It is known that C3NeF is not an exclusive autoantibody of this syndrome, it is also involved, and in a greater proportion, in other pathological situations not associated with lipodystrophy. Furthermore, many authors take it for granted that there is a pattern of synthesis of FD by subcutaneous tissue going progressively from more to less from the upper part of the body to the lower part, and that C3NeF destroys adipocytes in areas where more FD is synthesized; however, this has not been proven. Therefore, there must be other unknown pathophysiological factors which intervene in the clinical presentation, or the C3NeF in these patients has some different biochemical characteristics to those of all other patients without lipodystrophy.

Pathogenic mechanism of C3NeF in acquired partial lipodystrophy. C3NeF is uses the complement proteins synthesized by the adipocyte, leading to uncontrolled activation of the alternative pathway in the cell's surroundings. This situation progresses until the activation of the terminal pathway and the formation of the membrane attack complex on the surface of the adipocyte causing the lysis of the adipocyte.
Acquired partial lipodystrophy does not have a clear genetic basis. Reported cases are sporadic, with no Mendelian inheritance pattern. With some exceptions, it is most common to find families with one affected descendant, where the parents and other siblings are healthy. One very interesting case, reported in Spain, was that of a family in which two HLA-identical sisters presented with acquired partial lipodystrophy, recurrent infections, complement alterations and nephropathy26; however, a whole-exome sequencing analysis was not performed to find any common genetic markers related to the absence of adipose tissue. Another study conducted by a British group described, for the first time, a family with acquired partial lipodystrophy and C3G, in which the former segregates with the presence of C3NeF and the latter with a mutation in the CFH gene. The exome analysis did not reveal any common genetic variant among all the members affected by lipodystrophy.27
Susceptibility to developing acquired partial lipodystrophy has been associated with heterozygous mutations in the gene which encodes for the nuclear envelope protein called lamin B2 (LMNB2; OMIM# 608709).28 The results in four patients showed that two of the four variants were also present in donors without the disease. Moreover, the fat loss pattern affected the knees, thighs and the buttock region, which is highly atypical in this lipodystrophy that mainly affects the head, neck, trunk and upper extremities and does not affect the lower extremities. None of the patients presented C3 hypocomplementaemia and C3NeF was not detected. Three had diabetes mellitus and all presented with hypertriglyceridaemia, which is very rare in acquired partial lipodystrophy. Finally, it is worth noting that there was no segregation of these variants in the family members. Therefore, without more data or additional evidence, it is highly unlikely that the variants in the gene LMNB2 can be associated with acquired partial lipodystrophy.
Treatment of patients with acquired partial lipodystrophy: autologous adipose tissue transplant and anti-complement therapyA guide was published recently detailing guidelines for the diagnosis and clinical management of patients with rare lipodystrophy syndromes.29 This document, which was compiled with the contributions of endocrinology societies from all over the world and other expert committees, is a highly valuable tool when dealing with patients with these conditions.
The therapeutic approach for this condition is based exclusively on cosmetic repair. The autologous adipose tissue transplant from unaffected areas is considered to be a safe and effective treatment for restoring facial contour and breast volume, as these tend to be the most affected regions in these patients.30 This treatment is justified since, as a result of adipose tissue loss, most individuals end up suffering from a severe psychological disorder associated with the alteration to their physical appearance. Experience in this regard is limited, which means that it is difficult to predict the duration that the graft may have or how many interventions are necessary to obtain an acceptable restoration; but the positive effects for patients are clearly demonstrated, which is why it is highly recommended to value this approach in all cases. During adolescence, the risk of suffering from psychological disorders, which may make their personal and social lives difficult once they are adults, increases. Experts therefore maintain that restoration with autologous adipose tissue should be considered at a very early age, if possible during childhood.29,30
Advances in the knowledge of how the complement system works have been highly relevant for understanding the role of the complement system in the development of numerous diseases. Its involvement, both directly and indirectly, in the pathogenesis of some diseases has opened up an avenue to investigate how to regulate the response of the complement system. There are currently some drugs with a highly proven efficacy in reducing or restoring the damage caused by the dysregulation of the complement system. One example is the therapeutic use of the antibody anti-C5, called eculizumab (Soliris®, Alexion Pharmaceuticals), in diseases mediated by the complement system, such as paroxysmal nocturnal haemoglobinuria or aHUS.31 There is a case of a 14-year-old patient with C3G (dense deposit disease) associated with acquired partial lipodystrophy whose kidney function improved after receiving treatment with eculizumab.32 Regarding C3G, the efficacy data of the treatment with eculizumab are highly heterogeneous and inconclusive, which has prompted the conduct of numerous clinical trials.31 In the above-mentioned case, it was demonstrated that C5 blocking is very useful when other more general treatments, such as corticosteroid therapy or plasmapheresis, are insufficient. However, there are no data in the literature demonstrating that anti-complement therapy has any benefit on adipose tissue or metabolic disorders (if there are any) in patients with acquired partial lipodystrophy.
ConclusionsLipodystrophies, both the inherited and acquired types, are extremely rare diseases. This means that they are a real enigma when a patient comes to a medical consultation. In Barraquer–Simons syndrome (acquired partial lipodystrophy), which this review focuses on, most patients present with C3 hypocomplementaemia due to the presence of C3NeF, a potent deregulator of the alternative pathway. For its part, C3NeF is associated in most cases with the onset of C3G.
It is currently perfectly well known that the lack of homeostasis of complement system activation, in this particular case due to the presence of C3NeF, causes a massive systemic production of C3 fragments (especially C3b) which are deposited in the glomerulus, causing damage which may lead to chronic kidney failure. When acquired partial lipodystrophy was first described, the key role of the complement system in the biology of adipose tissue was barely known. It is known that this system is very important in the maturation of adipocytes, so much so that adipose tissue is practically the only producer of FD in the body. However, although there is much evidence to demonstrate the benefit of this relationship, there are data which prove that the dysregulation of the alternative pathway is related to the loss of adipose tissue, although the reasons why this event develops are not fully understood.
The most advisable treatment is autologous adipose tissue transplant from unaffected areas mainly to restore physical appearance problems. It is crucial to carry out this intervention in the earliest stages, especially during childhood, to reduce associated psychological disorders. On the other hand, anti-complement therapy in these patients has been shown to be effective by blocking the harmful effects of the complement system in the kidney, but with no improvement regarding adipose tissue.
Key concepts- •
Acquired partial lipodystrophy (Barraquer–Simons syndrome) is an very rare disease which is characterised by the progressive loss of subcutaneous adipose tissue of the head, neck, trunk and upper extremities, without affecting the lower extremities.
- •
Most patients with acquired partial lipodystrophy have reduced levels of the C3 component of the alternative complement pathway linked to the presence of C3NeF.
- •
Twenty-five per cent of patients develop C3G in the medium-to-long term, most due to dense deposit disease.
- •
Anti-complement therapy does not reverse the loss of adipose tissue, although its use is valuable in cases where C3G develops.
This study was funded by the Instituto de Salud Carlos III Health Research Fund (PI15-00255), the Sociedad Española de Nefrología (Spanish Society of Nephrology) (Fundación SENEFRO Aid for Research, 2016) and CIBERER (Center for Biomedical Network Research on Rare Diseases) (ACCI-2015).
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Corvillo F, López-Trascasa M. Lipodistrofia parcial adquirida y glomerulopatía C3: la desregulación del sistema del complemento como mecanismo común. Nefrologia. 2018;38:258–266.



