
Lipodystrophy syndromes are a heterogeneous group of rare diseases characterised by selective loss of adipose tissue, which often leads to insulin resistance with a tendency to develop diabetes, dyslipidaemia, hepatic steatosis, acanthosis nigricans and hyperandrogenism.1 Acquired partial lipodystrophy (APL; OMIM: 608709), also known as Barraquer–Simons syndrome, is one of the most common, and predominantly affects women (4:1). It is characterised by a progressive loss of subcutaneous fat, usually starting in childhood,2 and progresses from the head down, affecting the face, upper limbs, trunk and abdomen. Unlike other types of lipodystrophy, insulin resistance and metabolic complications, apart from hepatomegaly (60%), are rare.1,2
In 10–20% of cases, APL is associated with autoimmune disorders, especially lupus. The most common complication (20%) is the development of membranoproliferative glomerulonephritis type 2, or dense deposit disease (DDD) approximately 8 years after onset of the disease. The pathogenesis of such renal involvement includes activation of the alternative complement pathway (ACP).3 Patients show decreased serum C3 levels (70%) and are positive for C3NeF (80%),2 an autoantibody capable of altering the ACP and in the case of APL, it is suspected to play a role in the destruction of fatty tissue.4
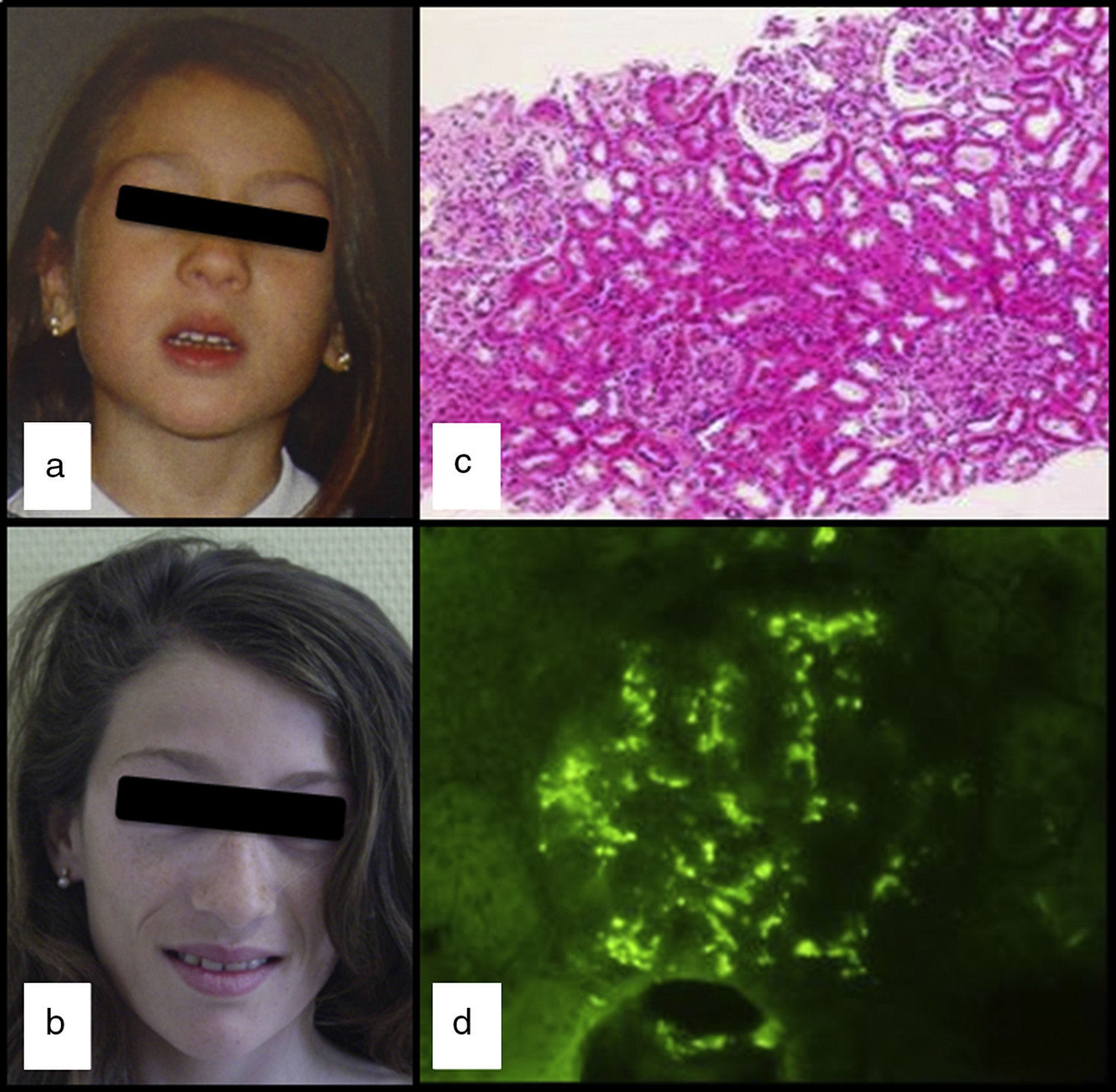
We had the case of a 15-year-old Caucasian girl with no relevant family or personal clinical history who was diagnosed of APL at the age of 5 due to progressive loss of subcutaneous fat from her face (Fig. 1a and b), with subsequent involvement of neck and shoulders. In her regular visits, no clinical or laboratory abnormalities had been observed, except for the slow progression of lipoatrophy and sustained decreased serum levels of C3 (26–48mg/dl; VN: 86–184). She had never had clinical or analytical data suggestive of nephropathy, dyslipidaemia, insulin resistance or hyperandrogenism. Her serum leptin (8.03ng/ml; NR: 15.3±8.1 SD) and adiponectin (8.3mg/ml; NR: 12.0±3.1 SD) levels were slightly low and the DXA body composition study showed a reduction in total body fat (17.7%; NR: 25.9±6.3).
 Progression of the loss of facial fat; (c) Glomeruli with increased mesangial cellularity, and focal and segmental involvement (H&E ×100) and (d) Mesangial granular immunofluorescence with anti-IgA.")
At the age of 13, after three days of fever and in the context of acute gastroenteritis, she was found to have macroscopic haematuria with non-nephrotic proteinuria (13mg/kg/day) and transient elevation of creatinine (maximum: 0.74mg/dl). The pyrexia subsided 24h later, but the macroscopic haematuria persisted for two weeks, with no other symptoms and with improvement in kidney function. Serum levels of IgA were found to be high (377mg/dl; NR: 40–350), C3 remained low and C3NeF negative. Studies of autoimmunity (anti-thyroid, anti-neutrophil and anti-nuclear antibodies) were negative. Percutaneous kidney biopsy was performed one month after the haematuria resolved. The specimen contained 14 glomeruli and revealed the presence of focal and segmental mesangial hypercellularity (Fig. 1c): M1, E0, S0 and T0 according to the Oxford classification5 (mesangial hypercellularity [M], endocapillary proliferation [E], segmental glomerulosclerosis [S] and interstitial fibrosis with tubular atrophy [T]). Only one of the glomeruli had a crescent that occupied 26–50% of the glomerulus. Immunofluorescence showed granular mesangial IgA and C3 deposits (Fig. 1d). Electron microscopy confirmed the presence of electron-dense deposits in the glomerulus, ruling out DDD. The patient was diagnosed with IgA nephropathy (IgAN), achieving clinical remission with spontaneous disappearance of the proteinuria and haematuria, but maintaining decreased serum levels of C3 (33mg/dl) and increased serum IgA (353mg/dl).
APL is a rare disease of uncertain aetiology. The association of APL with autoimmune diseases and DDD, in addition to the decrease in C3 and C3NeF positivity point to an autoimmune basis2; although a possible genetic predisposition has also been suggested.6 IgAN, meanwhile, is the most common glomerular disease in the world.7 The aetiopathogenesis of IgAN, which is not fully understood, also involves complement activation through the ACP7–9 and sometimes through the lectin pathway.10 Diagnosis is histological and kidney biopsy shows mesangial immune deposits of IgA1 with C3 and occasionally IgG or IgM. In the mesangial deposits, it is common to find components of the ACP (C3 and properdin), but not of the classic pathway (C1q and C4) which, taken together with normal serum C3 and other complement components, suggests that the complement activation occurs in the kidney itself.
To summarise, we present the first reported case of APL associated to IgAN. Although, given the incidence of IgAN, it could be a coincidence, the fact that the APL is often associated with nephropathy, the DDD, whose pathogenesis, like IgAN, involves activation of the ACP, raises the possibility of a common link between the two diseases.
Conflicts of interestThe authors declare that they have no conflicts of interest.
The authors thank Dr Fernando Corvillo, of the Immunology Department, Hospital La Paz, IdiPAZ, Madrid, for the study of C3Nef.
Please cite this article as: de Lucas-Collantes C, Pozo-Román J, Aparicio-López C, de Prada-Vicente I, Argente J. Lipodistrofia parcial adquirida (síndrome de Barraquer-Simons) y nefropatía IgA. Nefrología. 2016;36:556–558.



