
Although autosomal dominant polycystic kidney disease is the most common hereditary kidney disease, available data tend to be limited to after initiation of renal replacement therapy.
ObjectiveTo ascertain an overview of autosomal dominant polycystic kidney disease within the health area of Granada in southern Spain.
Materials and methodsFrom January 2007 to December 2016, we collected clinical, family and demographic information about all patients with autosomal dominant polycystic kidney disease, irrespective of whether or not they were treated with RRT, in the Granada health area. The computer software SPSS 15.0 and GenoPro were used.
Results50.6% of the 1107 diagnosed patients were men. 99.1% were Caucasian and 4–6 generations/family were studied. The geographical distribution was heterogeneous. There was no family history in 2.43%. The mean age of diagnosis was 34.0±17.80 years and the diagnosis was made after having offspring in 57.7% of cases. The main reason for diagnosis was family history (46.4%). The mean age of initiation of renal replacement therapy was 54.2±11.05 years. 96.3% of the deceased had some degree of renal failure at the time of death. The mean age of death was 60.9±14.10 years, the main cause of death being unknown in 33.5% of cases, followed by cardiovascular (27.8%).
ConclusionsCases and families were concentrated in certain geographical areas and a significant number of individuals were undiagnosed prior to cardiovascular death or diagnosed late after reproduction. Given that there is currently no curative treatment, the primary prevention strategy of preimplantation genetic diagnosis should play a leading role.
La poliquistosis renal autosómica dominante es la enfermedad renal hereditaria más frecuente aunque los datos disponibles generalmente son tras el inicio del tratamiento renal sustitutivo.
ObjetivoConocer la situación global de la poliquistosis renal autosómica dominante en el ámbito sanitario de Granada.
Material y métodosDesde enero 2007 hasta diciembre 2016 hemos recogido información clínica, familiar y demográfica de todos los pacientes con poliquistosis renal autosómica dominante, estuvieran o no en tratamiento renal sustitutivo, atendidos en el área de Granada. Se han utilizado los programas informáticos SPSS 15.0 y GenoPro.
ResultadosMil ciento siete pacientes diagnosticados, el 50,6% son varones. Se han estudiado 4-6 generaciones/familia. El 99,1% de raza caucásica. Hay áreas geográficas con mayor concentración. No hay antecedentes familiares en el 2,43%. La edad media de diagnóstico es de 34±17,8 años y en el 57,7% de los casos, el diagnóstico se produce después de tener descendencia. El principal motivo de diagnóstico son los antecedentes familiares (46,4%). La edad media de entrada en técnica es de 54,2±11,05 años. El 96,3% de los fallecidos tenían algún grado de insuficiencia renal en el momento del exitus. La edad media del exitus es de 60,9±14,10 años, siendo desconocida la principal causa de muerte (33,5%) seguida de la cardiovascular (27,8%).
ConclusionesCasos y familias se concentran en algunas áreas geográficas, un número importante de individuos están sin diagnosticar, fallecen antes por causa cardiovascular y se diagnostican tarde respecto al momento reproductivo. Dado que no hay tratamiento curativo, la estrategia de prevención primaria mediante el diagnóstico genético preimplantacional adquiere protagonismo.
Autosomal dominant polycystic kidney disease (ADPKD) is the most frequent hereditary kidney disease. The estimated prevalence is 1/800–10,001. The data on prevalence of ADPKD in Spain is not accurate and it is not known if there are variations in relation to periods of time.
Patients with ADPKD are 6–10% of the dialysis or kidney transplant population, so it is a disease with a great social impact.2,3 According to the Spanish Registry4 ADPKD is the sixth cause of renal replacement therapy (RRT). It is also the sixth cause of RRT in the Andalusia Region according to the information collected in the Information System of the Autonomous Transplant Coordination (SICATA).5 To date, data on this disease are available only from patients on RRT (transplant, hemodialysis [HD] and peritoneal dialysis [PD]). It is not known the overall magnitude of the problem, in the different provinces as well as at the national level since there are no records of those patients who are not on RRT. In 2016, the Spanish Society of Nephrology launched the national registry of ADPKD.
This disease begins from the moment of conception and evolves throughout life until it is diagnosed, usually by imaging tests performed if there is suggestive symptomatology. These patients present symptoms before the deterioration of renal function is evident (abdominal pains, hemorrhagic and / or infectious complications of the cysts and arterial hypertension [HTN]) which generates a demand of medical attention, in primary and specialized care which has not been quantified.
The main objective of our work is to analyze the epidemiology of ADPKD in our region to identify and design innovative health prevention strategies.
MethodsFrom January 2007 to December 2016 we have collected clinical, family and demographic information from all patients diagnosed of ADPKD, whether or not they were on RRT. For this we have collected information from a population covered by one health institution of the province of Granada and reference area: Documentation Services of San Cecilio and Virgen de las Nieves Hospitals in Granada, the records of the nephrology services, the HD centers arranged with the Andalusian Health Service, as well as the Urology, Pediatrics, Digestive, Neurology, Cardiology, Internal Medicine and Genetics of both hospitals; also from primary care and of from doctors and hospitals with private practice. A Study Group dedicated to ADPKD (GEEPAD) was created to collect data, evaluate all aspects of the disease and enhance preventive measures. This is a multidisciplinary group of health professionals that directly or indirectly had some type of relation with ADPKD patients.
All patients were informed about the study and were asked to sign a consent. Genealogical trees were made and the data have been stored in the GenoPro software.
The demographic, family, genetic and clinical information, (a total 238 variables), was introduced in a database created with the statistical program SPSS. For this article we have used the following variables: place of birth, current address, sex, race, age of diagnosis, whether it was before or after having the first child, number of descendants, number of children affected and those not studied, reason of the diagnosis, route of disease transmission, genetic study, need for RRT, type of RRT, involvement of other organs and location, HTN, infections and patient's situation at the present time, cause of death and if there was any deterioration of the kidney function at that time.
The unified T criteria of Ravine-Pei6–8 was used to made diagnosis.
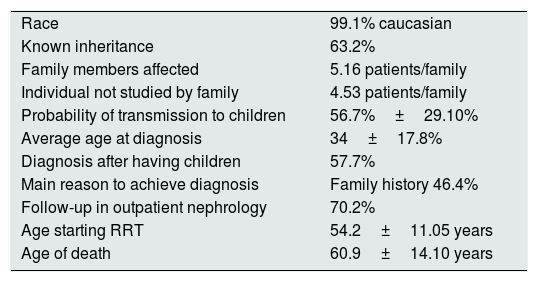
ResultsThere were 1107 patients with the diagnosis of ADPKD. The most relevant epidemiological information is shown in Table 1.
Main epidemiological results.
| Race | 99.1% caucasian |
| Known inheritance | 63.2% |
| Family members affected | 5.16 patients/family |
| Individual not studied by family | 4.53 patients/family |
| Probability of transmission to children | 56.7%±29.10% |
| Average age at diagnosis | 34±17.8% |
| Diagnosis after having children | 57.7% |
| Main reason to achieve diagnosis | Family history 46.4% |
| Follow-up in outpatient nephrology | 70.2% |
| Age starting RRT | 54.2±11.05 years |
| Age of death | 60.9±14.10 years |
The distribution by sex is homogeneous, 50.6% were males. A great majority of Caucasians (99.1%), blacks (0.5%) and gypsy (0.4%).
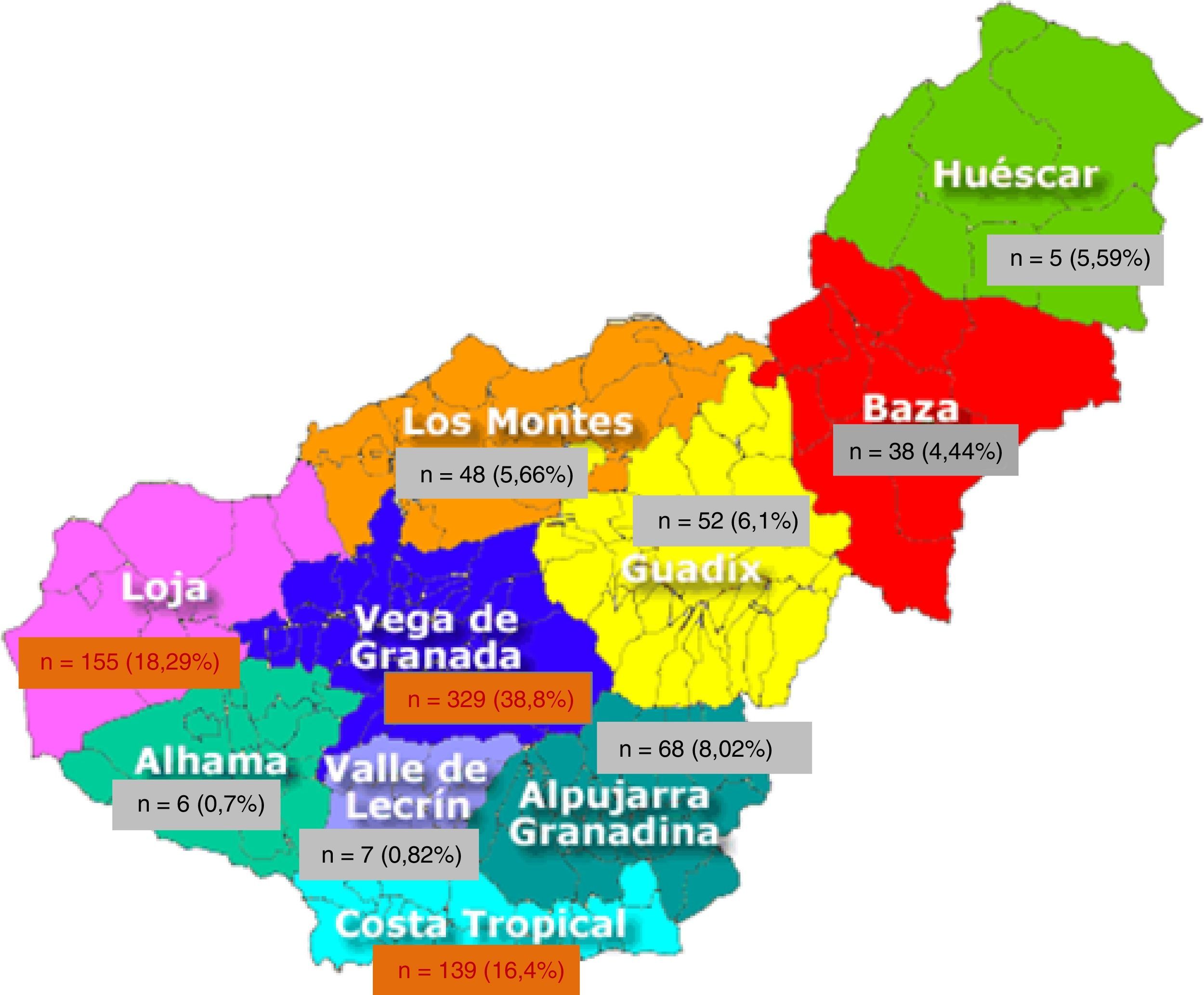
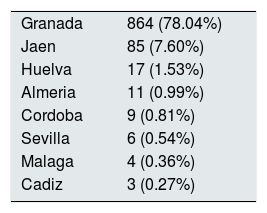
A 99.1% of patients were born in Europe and 97.9% in Spain, leaving 2.1% from other origin; 999 (90.24%) were born in Andalusia. By provinces, 78.04% (864 patients) were from Granada, followed by 7.6% from Jaen (Table 2). Of the 10 geographical areas of Granada, the most representative were Vega Granada, Loja and the Tropical Coast, where 56.2% of the total and 73.5% of the province were born (Fig. 1). Granada capital, Íllora and Motril were the 3 cities with largest number of patients: 13.7; 5.23 and 4.6%, respectively.
.")
In 63.2% of the cases the inheritance was clearly defined: 33.4% maternal, 29.8% paternal. We did not identify family history in 2.43% of patients. In 34.37% the inheritance route was not clearly identify: (a) doubtful maternal in 2.7%, (b) doubtful paternal in 4.69% and (c) unknown, without being able to rule out a family member affected in 26.98%
A total of 986 individuals (89.06%) were grouped in 251 families using family trees, after collecting information up to 4–6 generations. In patients >25 years, 77.3% have offspring, with an average of children of 1.9±1.60. The family impact of the disease, measured as the average of people with the disease within the same family, is 5.16 patients/family (range 1–41). The average of people not studied within the same family was 4.53 (range 0–38). In our series, the probability of transmission to children was 56.7% (range 0–100%), without differences in relation to gender.
The age of diagnosis of the disease was 34±17.8 years (n=641). In 57.7% of the cases the diagnosis was made after having offspring, in one case a mother was diagnosed during the first pregnancy.
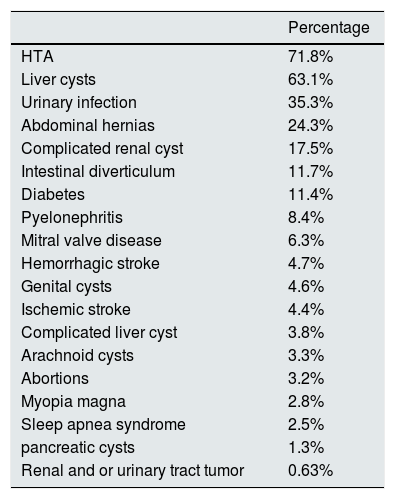
The main reason for diagnosis is family history in 46.4% of cases, followed by HTN (8.9%), chronic kidney disease (8.6%), abdominal-lumbar pain (7.4%), kidney stones (6.8%) and hematuria (4.6%). Among the extrarenal manifestations, the most frequent was hypertension (71.8%) followed by hepatic cysts (63.1%). The rest of the extrarenal manifestations are shown in Table 3.
Extrarenal manifestations and associated diseases.
| Percentage | |
|---|---|
| HTA | 71.8% |
| Liver cysts | 63.1% |
| Urinary infection | 35.3% |
| Abdominal hernias | 24.3% |
| Complicated renal cyst | 17.5% |
| Intestinal diverticulum | 11.7% |
| Diabetes | 11.4% |
| Pyelonephritis | 8.4% |
| Mitral valve disease | 6.3% |
| Hemorrhagic stroke | 4.7% |
| Genital cysts | 4.6% |
| Ischemic stroke | 4.4% |
| Complicated liver cyst | 3.8% |
| Arachnoid cysts | 3.3% |
| Abortions | 3.2% |
| Myopia magna | 2.8% |
| Sleep apnea syndrome | 2.5% |
| pancreatic cysts | 1.3% |
| Renal and or urinary tract tumor | 0.63% |
Out of 1107 registered patients, 642 are alive (536 live in Granada) being treated and followed in our healthcare setting: 70.2% in outpatient clinics, 23.5% in renal transplantation clinic, 4.8% in HD and 1.1% in PD. Of these, 246 (149 males and 97 females) are in an adequate reproductive age but 50% do not have children.
The mean age fro the initiation of RRT is 54.2±11.05 years. HD was started at 54.6±11.2 years old, PD at 51.8±7.7 years and kidney transplant was performed at 52.2±8.50 years.
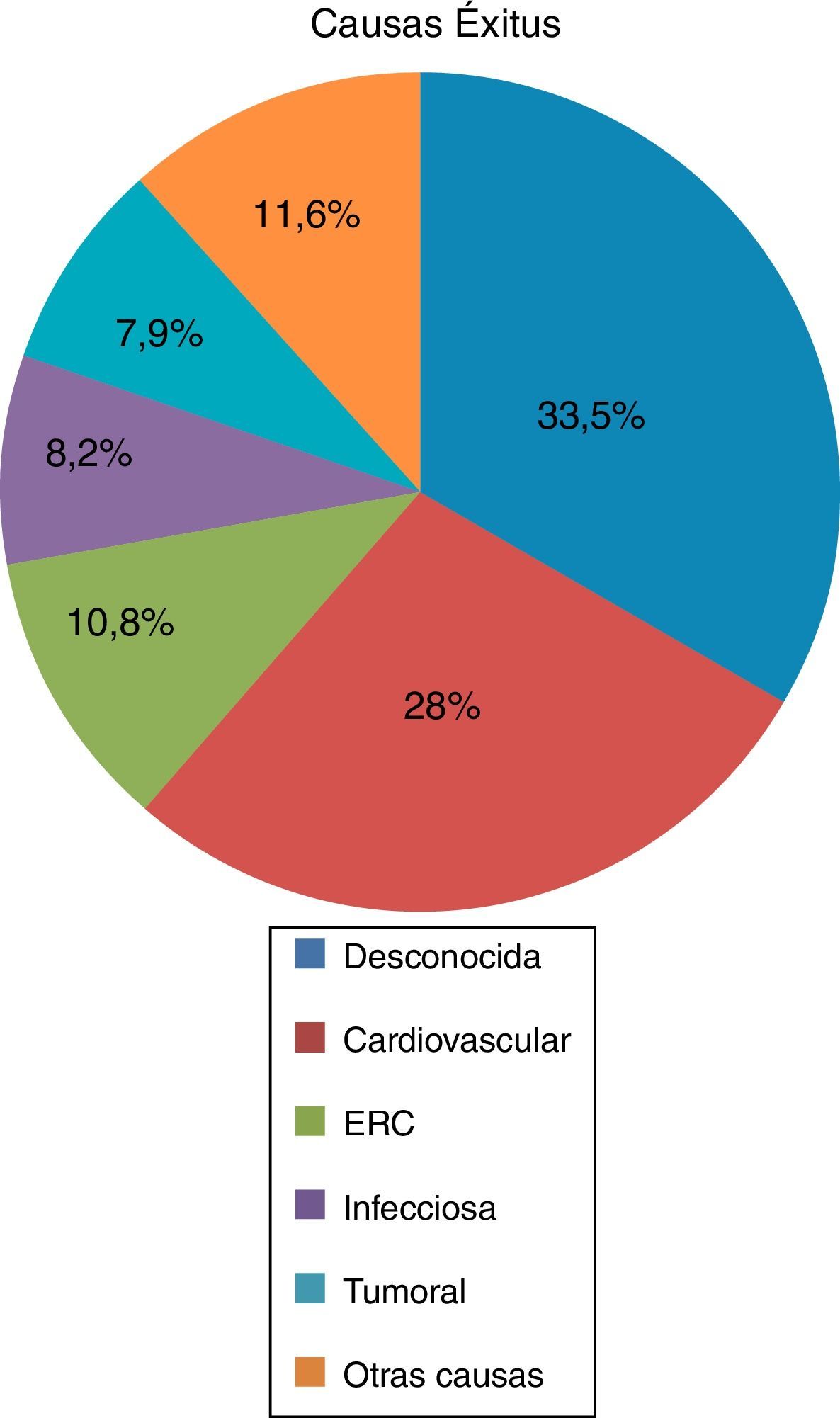
Of the deceased patients (n=350), 55.1% had received RRT in any of its 3 modalities, HD (53.9%) kidney transplantation (17.6%) and PD (4.6%). A 44.9% of the deceased patients did not receive RRT, although 96.3% of them had some degree of renal failure at the time of death. The average age of death was 60.97±14.14 years, the main cause of death was unknown (33.5%) followed by cardiovascular (27.8%), renal failure (10.8%), infectious (8.2%), tumor (7.9%), and other causes in 11.6% (Fig. 2).

Genetic evaluation was performed in at least one member in 58 families, whose results are the objective of another study. To summarize, mutations in PKD1 were the most frequent (84.5%), followed by those of PKD2 (6.9%) and PKD1 & PKD2 (3.4%). In 5.1% we found no detectable mutation or were variants without evidence of being pathogenic. Presently, the most striking and frequent mutation is c.10527_10528delGA ((p.Glu3509Aspfs * 117) a deletion located in exon 35 of the PKD1 gene, which causes a reading error and stop codon, causing shortening of polycystin 1. It is presented, so far in 6 families (121 affected people), from the region of Loja, in the area of Montefrío and Íllora, which we have not been able to relate to other family members by means of family trees, we call this the Loja mutation. Still, there are several families from the same area that have not been evaluated.
DiscussionOur study was performed to better understand the magnitude, clinical form of presentation and distribution of the ADPKD in our region in order to adequately address the needs of this population of patients.
Today, the values of prevalence of ADPKD are not accurate and varies greatly from one study to another. Iglesias et al.1 shows a prevalence of ADPKD in Western countries of one case/800–1000 inhabitants (between 10 and 12.5 cases/10,000 inhabitants) and, more recent studies in Europe establish a prevalence of 5–10 cases/10,000 people.9–11 According to our current data, with 536 patients living in the province with a population of 915,392 inhabitants (according to data from the National Institute of Statistics) the estimated prevalence in the health area of Granada would be 1:1707, or 5.85 cases/10,000 inhabitants. But this figure may be underestimating the true prevalence of the disease in our health area. Among the 251 families with a family tree analyzed there is an average of 4.53 members that have not been evaluate. This account for a total of 1155 individuals. Subtracting deceased individuals, those who do not live in Granada and considering a 50% probability of suffering the disease, there would be approximately 300 individuals affected without medical follow-up. Adding these patients to those already registered, the number of affected patients may increase to 836 and the estimated prevalence would be 1:1.094, a value in agreement with what is being described in the literature.
We have shown that we are facing a disease of heterogeneous geographical distribution; there are areas where the disease is more concentrated and others where it is practically symbolic. The knowledge of these areas allows us to develop strategies directed toward a more efficient distribution of health resources.
Routine ultrasound screening of asymptomatic children and adolescents with an affected father is still controversial because there is no effective treatment and it is already known that a normal ultrasound result does not exclude the disease in the early stages of life.8 However, we believe that it is important to inform families and offer the possibility of performing ultrasound screening of the disease in all members of the family if a parent is affected, even in asymptomatic children and adolescents. This strategy is appropriate because of the following reasons:
- 1.
Achievement of primary prevention of the disease, that is, prevent the transmission to future generations.
- 2.
Perform secondary prevention. Understanding by this term the implementation of all current means to slow the progression of the disease and decrease cardiovascular risk.
According to a survey conducted on ADPKD by a pharmaceutical company in Spain in 2014,12 44% participants decided not to have children after the diagnosis of the disease. And according to another survey conducted in the United Kingdom in 2014, 59% of patients with ADPKD would choose preimplantation genetic diagnosis (PGD) if it were available in the portfolio of Social Security Services.13
Something relevant of our study is that the average age of diagnosis of the disease in our province is 34 years and in more than half of the cases already had. Therefore, we must anticipate the diagnosis to have primary prevention. Spanish health regulation support this strategy.14 The same strategy was adopted in the Andalusian region15 and in the Province of Granada.16
The Spanish Clinical Guidelines of ADPKD recommend that affected patients inform their first-degree relatives about the risk of suffering from the disease and consider that screening should be offered together with genetic counseling.17
The mortality rate of polycystic patients is almost 3 times higher than that of the general population18 being cardiovascular death a frequent cause of death, thus early diagnosis is important. By acting on cardiovascular risk factors, survival will improve and the progression of renal failure will slow down.
This disease should have a multidisciplinary approach involving various in-hospital specialties and primary care professionals. The later are closer to the families, perform the screening and refer the patient to specialists. Within the multidisciplinary group we believe that gynecologists, geneticists and pediatricians have an important role. The gynecologists, because they advise about the more appropriate reproductive method in each couple. Geneticists perform and interpret the mutational studies and provide adequate genetic counseling. Pediatricians, who are the ones who must be vigilant, so that children of parents with ADPKD can be diagnosed early and identify risk factors that require early intervention.19
ConclusionsThe obtained epidemiological data on ADPKD is fundamental to adequately address different aspect of the disease. In our region, the distribution of ADPKD is not homogenous, and it is concentrated in areas where health intervention may be necessary. We have observed a late diagnosis and in more than half of the cases after having offspring. In addition, there are still numerous undiagnosed individuals. We conclude that it is convenient to implement different level of information and screening program where the care management of the family tree is essential.
These patents die younger than the general population, with cardiovascular disease being the main cause of death. Early diagnosis along with a greater control of cardiovascular risk factors should improve life expectancy.
Our group defends the strategy of primary prevention based on establishing the diagnosis of the disease before having offspring, genetic counseling and promoting the use of reproductive techniques that prevent the transmission of the disease such as Preimplantation Genetic Diagnosis already available in the public health system.
Conflict of interestsThe authors declare that they have no conflicts of interest.
To patients and families with ADPKD.



