


Con el término de nefropatías intersticiales se denominan aquellas enfermedades que afectan de forma predominante al intersticio renal, aunque también se pueden encontrar dañados, en mayor o menor medida, todos los elementos integrantes del parénquima renal (glomérulos, túbulos y vasos). Dado que las células del túbulo renal suelen estar dañadas, algunos autores prefieren el término de nefropatías túbulointersticiales (NTI) [1]. El daño túbulointersticial está claramente relacionado con la progresión de la enfermedad renal [2].
Dentro de las NTI, se conocen formas familiares con un perfil clínico muy heterogéneo, incluso dentro de la misma familia [3]. Dado que en varias familias se constataba la presencia de quistes corticomedulares, y se diferenciaban claramente por la edad de comienzo, se acuñó el concepto de "Complejo Nefronoptisis-Enfermedad Quística Medular" [4] [5]. Nefronoptisis define las formas infantiles con herencia autosómica recesiva, y los genes inicialmente descritos fueron NPHP1 (proteína Nefroquistina) y INVS (proteína Inversina) [6] [7], habiéndose descrito actualmente hasta 19 genes causantes de distintas formas de nefronoptisis [7]. El término Enfermedad Quística Medular se aplicó a las formas adultas con herencia autosómica dominante, y el primer gen identificado fue el UMOD (proteína Uromodulina) [8] [9].
El mayor conocimiento de los genes implicados, y la presencia muy inconstante de quistes, ha determinado la evolución de estos conceptos. Se espera que con ello, vayamos delimitando mejor el perfil clínico de esta entidad, de presentación anodina y de evolución variable [10].
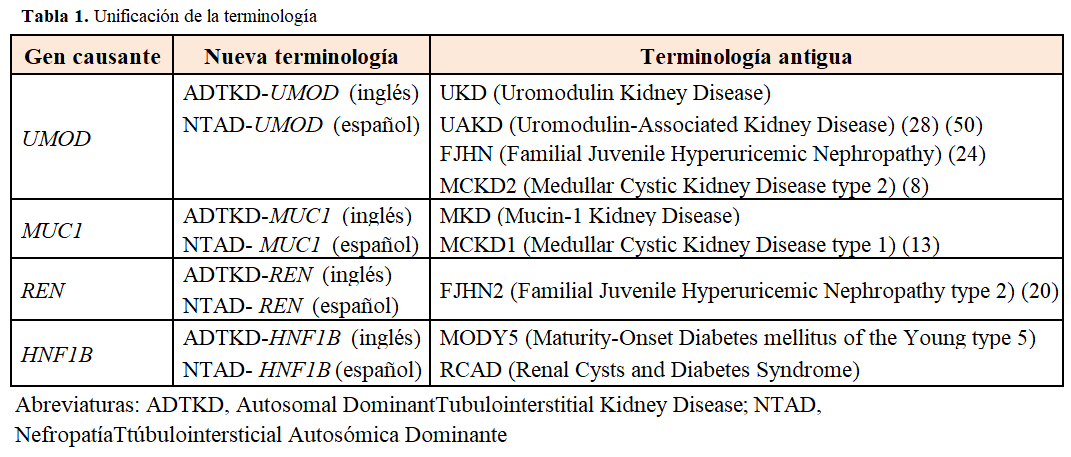
TerminologíaEl término Nefropatía Tubulointersticial Autosómica Dominante (NTAD) ha sido establecido por las guías KDIGO utilizando la sigla en inglés ADTKD [12]. Previamente a este consenso, la nomenclatura era muy variada con las confusiones clínicas que ello implica [10] [11] (Tabla 1).
El grupo de trabajo de las Guías KDIGO, decidieron unificar terminologías y características clínicas de estas enfermedades renales raras hereditarias [12], que tienen en común la fibrosis túbulointersticial y la progresión lenta a enfermedad renal crónica terminal (ERCT) [1] [13]. Las ventajas de la nueva terminología son:
• Refleja que se trata de enfermedades de origen genético con un patrón de herencia autosómico dominante. Esto conlleva la separación del resto de nefropatías tubulointersticiales crónicas de carácter adquirido, así como el grupo de las nefronoptisis (patrón de herencia autosómico recesivo).
• Resume las características clínicas de la patología causada por mutaciones en 4 genes diferentes.
• Permite la sospecha clínica en ausencia o antes de objetivar lesiones histológicas o genéticas.
• Permite diferenciarlas de otras enfermedades autosómicas dominantes de origen tubular (como es la poliquistosis renal autosómica dominante o la acidosis tubular distal).
• Evita terminologías previas que podrían causar confusión, sobre todo las que incluyen los términos “enfermedades quísticas o quistes medulares”.
• Es una terminología simple y fácil.
Las NTIAD son una de las tres causas más frecuentes de nefropatías monogénicas (junto con PQRAD y Alport). Se estima que globalmente las NTIAD suponen alrededor del 5% [14] [15].
Características clínicas generalesLa penetrancia es muy alta, cercana al 100% en los pacientes longevos, sin embargo, la severidad y la edad de aparición varía mucho tanto a nivel interfamiliar como intrafamiliar, y dependiendo del gen mutado [12] [16] [17].
La enfermedad renal es de lenta evolución y la edad de llegada a ERCT es muy variable, pudiendo oscilar entre los 25 y 70 años en pacientes con mutación en el gen UMOD [3] [18] [19]. En la cohorte española de NTAD se objetivó una edad media de ERCT de 51 años en los pacientes con NTAD causada por mutaciones en el gen MUC1 y de 56 años en los pacientes con NTAD causada por mutaciones en el gen UMOD (P=0.1), así mismo, los pacientes con mutación en el gen MUC1 presentaban mayor riesgo de desarrollar ERCT (HR, 2.24; P = 0.03) [17]. La tasa de declive del filtrado glomerular es muy variable también en función del gen mutado pero suele tratarse de una insuficiencia renal de lenta evolución [3] [16] [17] [20].
Los signos y síntomas acompañantes son muy inespecíficos.
• La proteinuria es negativa o es de poca cuantía (<1g/día) [3] [16] [17] [20].
• El sedimento urinario suele ser normal o excepcionalmente con microhematuria [3] [16] [20].
• El tamaño renal es normal y se reduce a medida que progresa la enfermedad. Los quistes renales preferentemente corticomedulares, son relativamentes frecuentes pero inconstantes y suelen encontrarse en fases avanzadas de la enfermedad [3] [16] [17] [20].
• En los pacientes con mutación en el gen UMOD se han reportado edades de aparición de la hiperuricemia muy variables, desde los 3 hasta los 51 años [3] [18] pero tienen una prevalencia mucho mayor de hiperuricemia que el resto de NTAD causada por otros genes [17].
• La hipertensión es un hallazgo habitual pero no aparece precozmente ni suele ser grave [12].
• La anemia suele ser precoz y desproporcionada al grado de insuficiencia renal, siendo más grave cuando la enfermedad está causada por mutación en el gen REN [21].
A pesar de tratarse de una afectación tubulointersticial, las anomalías funcionales tubulares no son características, sólo se han descrito defectos en la concentración de la orina o defectos en la reabsorción de ácido úrico a nivel del túbulo proximal, en relación a las vaiantes patogénicas de UMOD fundamentalmente En estudios experimentales se han objetivado otros transportadores catiónicos dañados, pero no hay evidencias claras en la repercusión clínica de los pacientes [22] [23] [24].
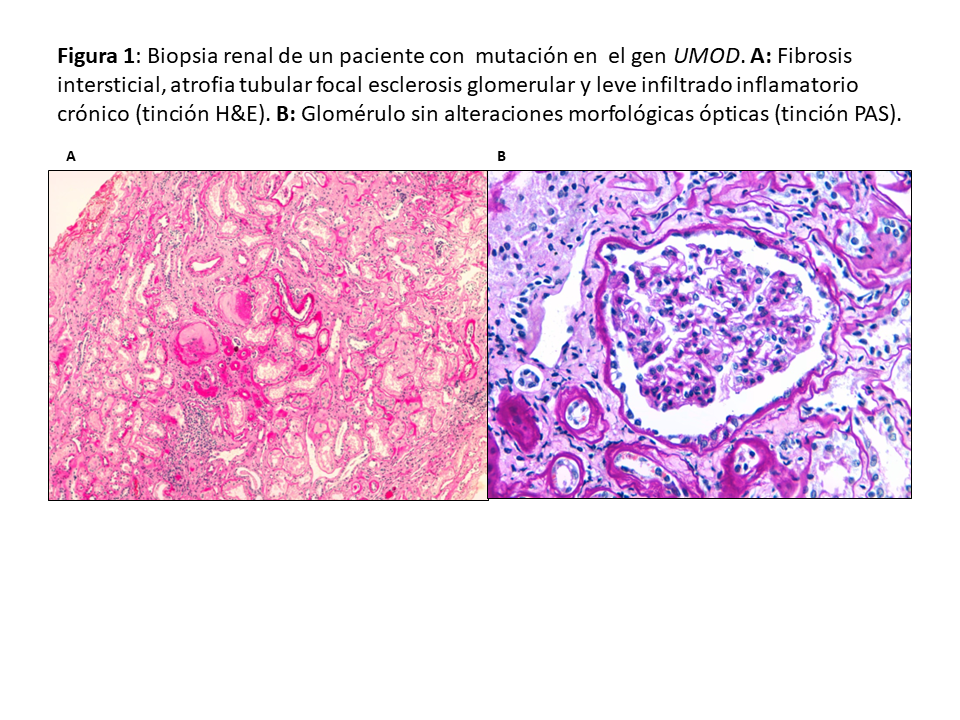
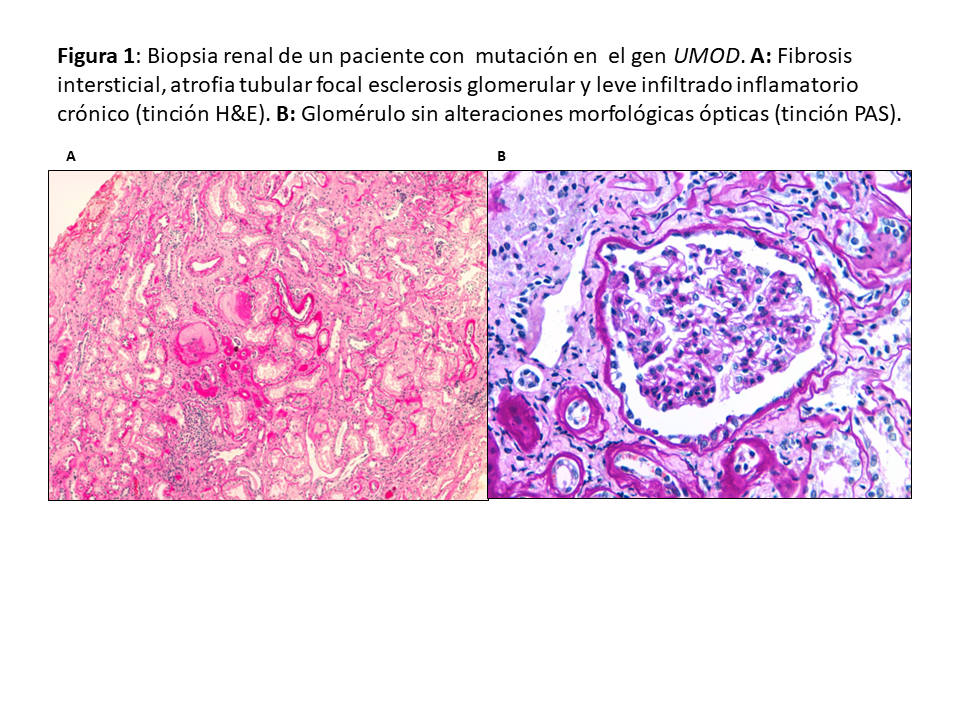
Patrón histológicoDebido a su poca expresividad clínica y lenta evolución, el diagnóstico por biopsia renal es infrecuente. Tienen en común fibrosis intersticial con atrofia tubular y glomérulos normales (Figura 1). Es frecuente la pérdida progresiva de la membrana basal glomerular y puede observarse dilatación tubular y microquistes tubulares [25] [26] [20] [16] [3]. La inmunofluorescencia (IF) es negativa. La microscopía electrónica no suele aportar mucho al diagnóstico diferencial aunque puede observarse acumulación de la uromodulina mutante en el retículo endoplásmico de las células de la rama ascendente gruesa del asa de Henle [27].
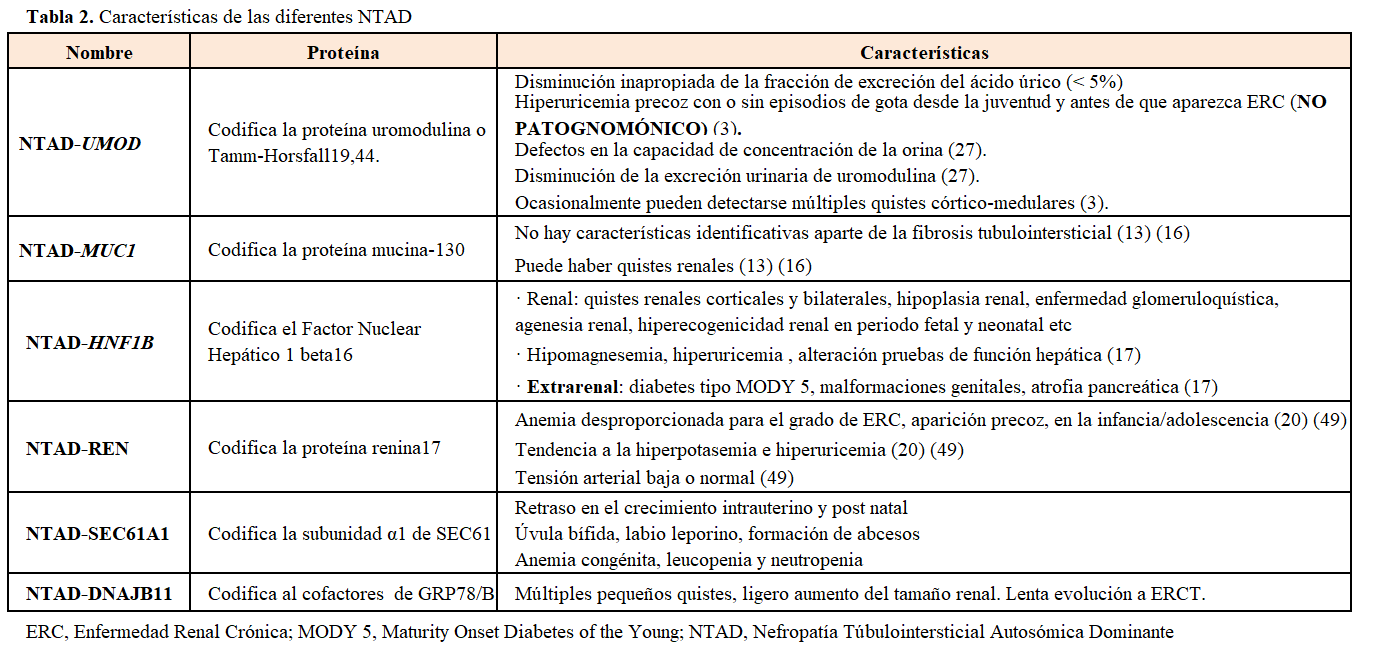
Defectos genéticos y presentación clínicaHasta la fecha se han descrito 6 genes causantes (Tabla 2). Aunque el porcentaje de NTAD causada por variantes patogénicas en cada uno de estos genes no está claramente definida, las variantes patogénicas en UMOD y en MUC-1 darían lugar a un mayor porcentaje de casos de NTAD que los causados por variantes patogénicas en los genes REN o HNF1b [12]. En la cohorte española la principal causa de NTAD fue la variante patogénica de MUC1 c.(428)dupC seguida de las variantes patogénicas del gen UMOD [17].
Otra cohorte irlandesa publicada ren 2019 que estudió a 16 familias mostró resultados similares, la variante patogénica en MUC1 fue la más frecuente seguida de cerca por la variante patogénica en UMOD. En algunos individuos el diagnóstico se realizó por detección de proteína anómala (MUC1fs) en orina [28].
• NTAD causada por variantes patogénicas en el gen UMOD:El gen UMOD (región cromosómica 16p2) codifica para la proteína uromodulina o de Tamm-Horsfall, la proteína más abundante de la orina, cuya función no ha sido totalmente aclarada (se ha relacionado con la impermeabilización del túbulo distal y con una actividad proinflamatoria) que se produce en las células epiteliales de la porción ascendente del asa de Henle [23] [29] [30]. La uromodulina mutante se acumula en el retículo endoplásmico de las células y disminuye su liberación y excreción urinaria [30]. Así mismo, se inhibe el tráfico intracelular del cotransportador de Naþ-Kþ-2Cl_ NKCC2 a la superficie apical de la rama ascendente gruesa de Henle, disminuyendo la reabsorción de Na y como consecuencia se produce una depleción de volumen que favorece la reabsorción proximal de ácido úrico [31].
Se han descrito más de 130 variantes patogénicas en el gen UMOD, la gran mayoría missense, en los exones 3 y 4 [15] [32].
Clínica: cursa frecuentemente con hiperuricemia y a veces con crisis gotosas, debido al descenso de la excreción fraccional de ácido úrico [33]. La hiperuricemia es desproporcionada para el grado de insuficiencia renal, puede precederla y aparecer en la infancia y adolescencia [3]. La enfermedad renal terminal suele ocurrir entre los 25 y 70 años y más precozmente en los que padecen gota [25]. Estos datos se confirman en una cohorte internacional reciente de 722 pacientes donde se describió mayor riesgo de progresión hacia ERCT en hombres y también se asoció con la llegada precoz a ERCT la aparición temprana de gota [34]. Algunos estudios han demostrado enlentecimiento en la progresión de la enfermedad renal (estadio 3-4) en pacientes con hiperuricemia tratados con febuxostat frente a placebo [35]. No está claro si el alopurinol enlentece la progresión de la insuficiencia renal [1] [36].
Se han descrito casos, en fases avanzadas de la enfermedad, con proteinuria subnefrótica que pueden ser erróneamente diagnosticados de HFS secundaria [37].
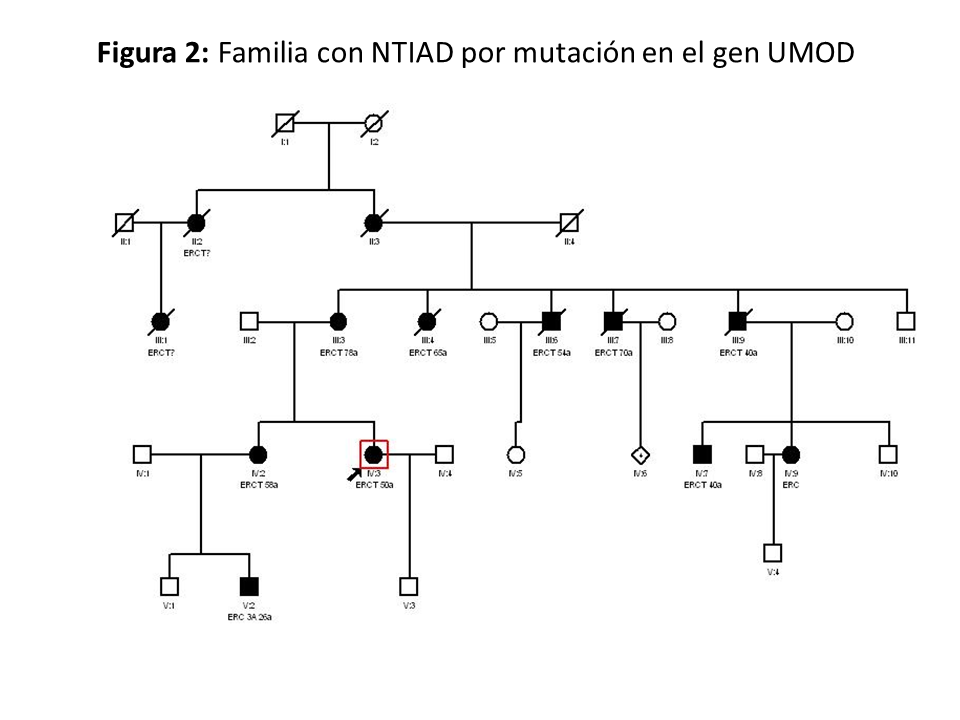
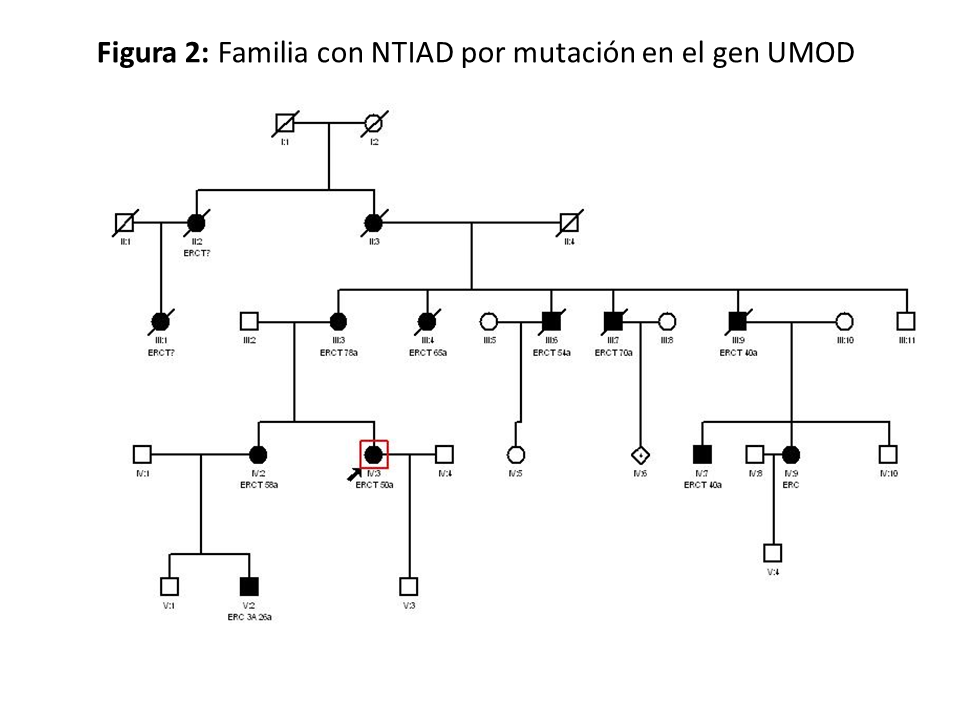
En la (Figura 2) se muestra un ejemplo de familia con NTAD por mutación en el gen UMOD, donde se observa la gran variabilidad intrafamiliar.
• NTAD causada por variantes patogenéticas en el gen MUC1:El gen MUC1 (región cromosómica 1q22) fue identificado como causante de NTAD en 2013 [38] aunque estudios de ligamiento habían identificado el locus candidato en el cromosoma 1q21 en 1998 [39]. En 2002 se describió la primera familia afecta en Chipre [26]. Este gen, de 7 exones, codifica para la mucina-1, una proteína transmembrana altamente glicosilada con gran expresión en la nefrona distal. Las mucinas tienen una función protectora de la barrera epitelial luminal. La variante patogenética mayoritaria consiste en la duplicación de una citosina de una cadena de 7 citosinas que se encuentran en número variable (20-125) de 60 pares de bases repetidos en tandem (VNTR) de unidades del gen. Otros cambios patogénicos incluyen la adición de un residuo de guanosina o la pérdida de 2 residuos de citosina. Se produce en todos los casos una mutación de cambio de pauta de lectura (frameshift) resultando una proteína alterada (MUC1-fs). Esta se acumula favoreciendo la apoptosis de las células tubulares epiteliales. Se desconoce el mecanismo por el que este producto del gen causa fibrosis tubulointersticial y por qué no se afectan otros tejidos donde la mucina-1 se expresa (mama, pulmón, glándulas sebáceas y salivares) [40].
Sin embargo, se están produciendo avances importantes. En este sentido, hay varios trastornos genéticos incluida la NTIC- MUC1 que parecen tener en común la acumulación de proteínas que se produce por alteración en el plegamiento causando un “atasco” molecular. Recientemente se ha identificado una molécula, la BRD4780 que aclararía la MUC1-fs (acumuladas en vesículas, unidas a receptores TMED9) y podría ser un punto de partida para el desarrollo de fármacos [41], no sólo en esta nefropatía sino en otras proteinopatías tóxicas, enfermedades heterogéneas que comparten mecanismos similares [42].
Se ha identificado a la mucina-1 como factor protector del COVID-19. En alguna serie se ha descrito que los pacientes con NTIAD-MUC1 tienen más del doble de riesgo de padecer Covid y una peor evolución comparados con NTIAD-UMOD [43]; la indicación de vacunación en estos pacientes está, si cabe, más recomendada y especialmente en aquellos pacientes con trasplante renal.
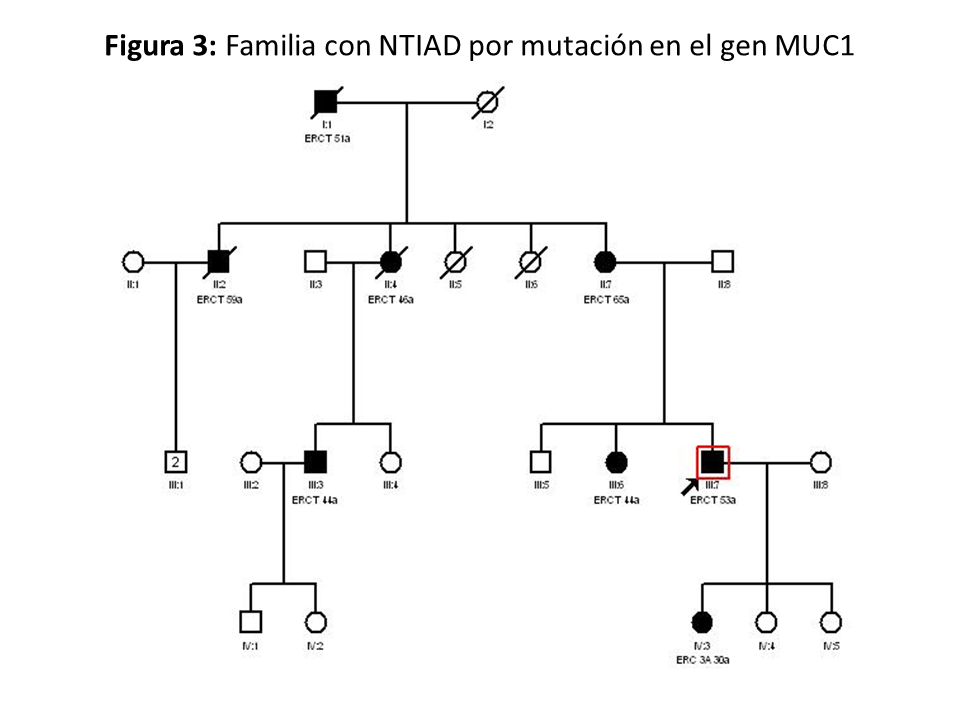
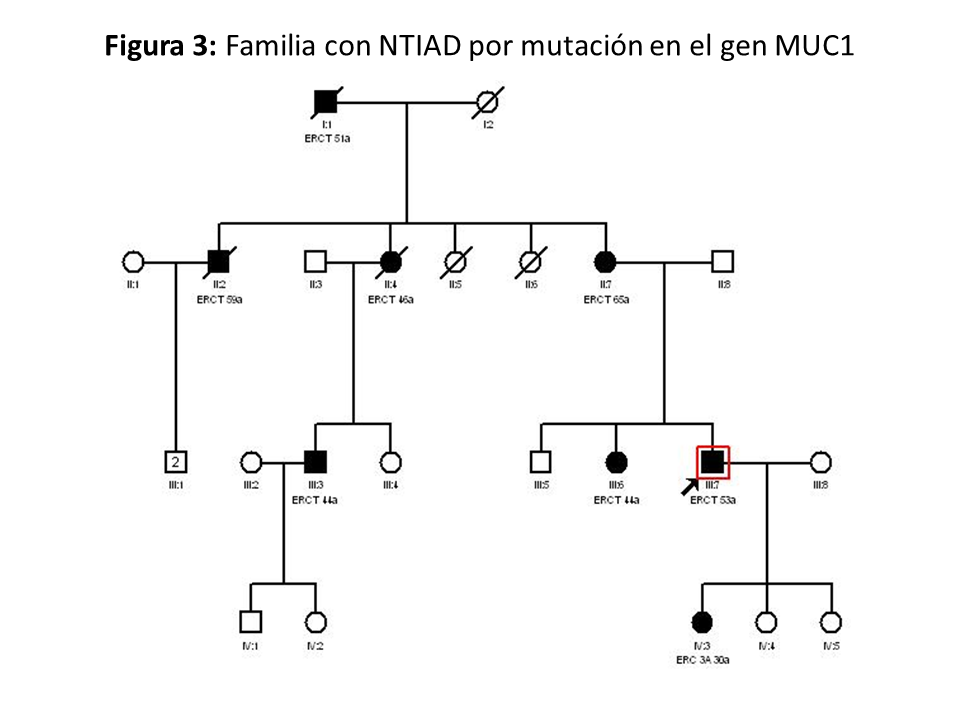
Clínica: Los casos descritos de pacientes con variante patogénica en el gen MUC1 no muestran ninguna manifestación extrarrenal o anomalía clínica característica salvo la fibrosis tubulointersticial [16] [17]. En la (Figura 3) se muestra el ejemplo de otra familia con NTAD por mutación en el gen MUC1.
• NTAD causada por variante patogénica en el gen HNF1B:El gen HNF1B (OMIM 17q12 and 189907) codifica para el factor nuclear hepático 1b (HNF1b), un factor de transcripción que regula múltiples genes expresados durante la embriogénesis de diferentes tejidos (renal, hepático, pancreático o genital) [44] [45]. El gen HNF1B regula a su vez el gen UMOD y varios genes implicados en la poliquistosis renal autosómica dominante [46].
Clínica:Las variantes patogénicas del gen HNF1B pueden causar múltiples alteraciones extrarrenales y sólo una minoría dan lugar a nefropatía tubulointersticial [47] [48]. Sólo se incluyen como NTAD aquellas formas clínicas en las que la fibrosis tubulointersticial es la principal manifestación [12] [20] [44]. Los primeros síntomas pueden presentarse en la infancia e incluso en el periodo prenatal.
• NTAD causada por variantes patogénicas en el gen RENEl gen REN (OMIM 1q32.1 and 179820) codifica la proteína renina. El precursor, preprorrenina, se deposita en las células del túbulo renal y se produce apoptosis celular y también acumulación intracelular de la renina anómala [21]. La renina además se expresa en las células tubulares de la nefrona distal solapándose con los patrones de expresión tubular de otros genes de NTAD [49].
Clínica:Las características clínicas de la enfermedad pueden atribuirse a la hipoactivación del sistema renina angiotensina aldosterona. Se manifiesta con renina y aldosterona baja, tendencia a hiperpotasemia y cierto grado de hipotensión. Como consecuencia, tienen mayor riesgo de IR en situaciones de depleción volumétrica o uso de AINES [50] [51]. La anemia precoz, desproporcionada al grado de insuficiencia renal, puede incluso aparecer en la infancia, desaparecer en la adolescencia, y reaparecer en fases más avanzadas de la enfermedad renal [21].
Recientemente se ha publicado una cohorte de 111 individuos de 30 familias con variantes heterocigóticas en gen REN.
En función de la región afecta de renina (región péptido señal, prosegmento o péptido maduro), las manifestaciones variaban de mayor gravedad y presentación precoz en la infancia (las más frecuentes) a fenotipo de afectación más tardía, con gota frecuente y sin anemia (similar a la clínica de NTAD-UMOD) [52].
NTAD causada por variantes patogenéticas en el gen SEC61A1El gen SEC61A1 (OMIM 3q21.3 and 609213) codifica para la subunidad α1 de la proteína transportadora SEC61, un canal translocador del retículo endoplasmático. Las mutaciones en este gen producirían una proteína anómala que desestabilizaría el poro, afectando a proteínas transmembrana y secretoras, entre ellas la uromodulina, la mucina 1 y la renina. La mutación de SEC61A1 también causaría problemas en la homeostasis del calcio y en el metabolismo energético de la célula [53] [54].
Clínica:Se han descrito 2 familias con mutación en este gen. Algunos de los miembros presentaban anemia congénita, y retraso en el crecimiento intrauterino y postnatal, y también hay descritos algunos miembros con úvula bífida, labio leporino o presencia de abcesos cutáneos [53] [54].
• NTAD causada por variantes patogénicas en el gen DNAJB11El gen DNAJB11 (OMIM 3q27.3 618061) es un cofactor de una chaperona en el RE (retículo endoplasmático) que controla el plegamiento, el tráfico y la degradación de proteínas. Se han descrito cambios patogénicos, la mayoría truncantes, que dan un fenotipo híbrido entre PQRAD y NTIAD.
Clínica:Se suele manifestar como pequeños quistes múltiples con ligero aumento del tamaño renal y en la mitad de casos con quistes hepáticos. Presenta evolución lenta hacia ERCT entre la 6ª-7ª década de la vida [14] [15].
Criterios de sospecha diagnósticaLos criterios de sospecha, establecidos por las guías KDIGO de NTAD [12] se describen en la (Tabla 3).
La NTAD también debería ser considerada en pacientes aislados, sin antecedentes familiares conocidos, pero que presenten las características previamente mencionadas. Podrían tratarse de casos de novo o de diagnósticos no realizados o erróneos en otros familiares. Estos casos deberían considerarse como “sospechosos de NTAD” si no hay una histología compatible en la biopsia renal [12].
El análisis genético es el único método para confirmar definitivamente el diagnóstico y poder clasificar el tipo de NTAD, así como de excluir la enfermedad en aquellos miembros de la familia que no tengan mutación en el gen causante. Sin embargo, es importante reseñar que un resultado genético negativo en una familia, no excluye totalmente el diagnóstico, ya que no todos los genes causantes de NTAD han sido identificados y, además, las técnicas de diagnóstico genético no tienen una sensibilidad del 100%. Hoy en día las técnicas de secuenciación masiva de genes resultan de gran ayuda ya que permiten, ante la sospecha de una NTAD, analizar la mayoría de los genes descritos, exceptuando la mutación patogénica de MUC1 El análisis de este gen es mucho más complejo y se realiza sólo en algunos centros especializados [38].
Dicho estudio no puede ser realizado simplemente por secuenciación de Sanger o masiva, ya que la única mutación MUC1 conocida hasta el momento se localiza en una región de repetición en tándem rica en guaninas-citosinas (GC-rich 60-base VNTR). Otros cambios patogénicos incluyen la adición de un residuo de guanosina o la pérdida de 2 residuos de citosina. Todas las variantes patogénicas resultan en la síntesis de una proteína anómala (fs) que termina prematuramente [14]. A pesar de los pocos casos descritos con mutación MUC1, se ha observado algún individuo de edad avanzada con esta mutación MUC1 que no presenta síntomas, por lo que podría presentar penetrancia incompleta (portadores de la mutación asintomáticos).
Además del perfeccionamiento de los análisis mutacionales para identificar la mutación MUC1, están emergiendo estrategias diagnósticas alternativas como la medición de niveles de uromodulina o detección por inmunohistoquimia de MUC1fs en orina [55].
Recomendaciones de estudio genéticoA pesar de que actualmente no hay ningún tratamiento específico para la NTAD, el estudio genético se sigue recomendando para el diagnóstico definitivo de la NTAD y de la identificación de los subtipos, así como para confirmar/descartar la enfermedad en los familiares asintomáticos o con clínica dudosa. En la (Tabla 4) se describen las situaciones en que se recomienda realizar el estudio genético secundario.
En los casos en los que se identifica la mutación causal, los pacientes deben recibir consejo genético sobre el riesgo de transmisión y también se debe ofrecer la posibilidad del estudio genético a otros miembros de la familia. En general, no se recomienda realizar el estudio genético a menores de edad dada la ausencia de opciones terapéuticas específicas, salvo si hay sospecha de mutación en gen REN, dado que se podrían beneficiar de tratamiento con EPO más precozmente y/o fludrocortisona [51].
Con los rápidos avances en el campo de la genética, los métodos más apropiados para el diagnóstico de estas nefropatías evolucionan. Actualmente, en caso de alta sospecha clínica se puede ir directamente a la secuenciación de un gen concreto, por ejemplo UMOD, pero en general se recomienda solicitar un amplio panel de genes dado que en muchos casos los fenotipos son heterogéneos y se pueden clasificar en muchas categorías [56] [57] [58] [59].
Es importante resaltar que la NTAD, aunque infrecuente, probablemente está infradiagnosticada por su clínica inespecífica y poco expresiva; y concretamente, por la dificultad de diagnóstico de la NTIAD-MUC1, ya no se puede detectar por NGS y su análisis sólo se realiza en uno pocos centros a nivel mundial [14].
Recomendaciones de seguimiento y manejo- Se recomienda el estudio genético en pacientes con sospecha de NTAD
- Se recomienda estudio genético de los familiares en riesgo
- En casos individuales de clínica sugestiva de nefropatía intersticial crónica, sin antecedentes familiares, el rendimiento del estudio genético es muy bajo. Primero, se debería pensar en otras causas de enfermedad renal.
- Se recomienda controlar los factores de riesgos conocidos (HTA, DM, tabaco, obesidad…) que puedan influir en el daño renal y realizar análisis anual de la función renal.
- En los menores en riesgo de enfermedad por mutación en UMOD o MUC-1, las opciones de tratamiento son escasas. Los niños en riesgo de tener una mutación en HNF1B o REN deben remitirse al nefrólogo pediátrico. En estos casos se beneficiarían de un manejo más precoz.
- Los AINES deben evitarse en todos los pacientes con NTAD y especialmente en aquellos con mutaciones en el gen REN, pues son más susceptibles a que empeore su función renal.
- No se recomienda la restricción de sal en pacientes con defectos genéticos en UMOD y REN; puede agravar la hiperuricemia.
- Se desconoce si la dieta pobre en purinas es beneficiosa en pacientes con mutaciones en UMOD.
- En general, los diuréticos deben usarse con precaución ya que pueden agravar la hiperuricemia y la depleción de volumen.
- Los pacientes con enfermedad asociada a la UMOD que desarrollan gota pueden tener nuevos episodios. Estos pueden prevenirse con el alopurinol y, si este no se tolera, con febuxostat una vez resuelta la primera crisis.
- No hay consenso de los beneficios del bloqueo del eje renina-aldosterona en la progresión de la nefropatía. Si se utilizan, se recomienda tratar con losartan a los que tengan hiperuricemia por ser el único uricosúrico.
- El tratamiento con ISGLT2, como en otras nefropatías, podrían ser útiles para retrasar la progresión de la enfermedad renal [43] [60].
- En la NTAD por defecto del gen REN, los pacientes suelen requerir EPO más precozmente y fludrocortisona para tratar los síntomas de hipotensión pero no debe usarse en aquellos con deterioro del filtrado glomerular, HTA, hiperpotasemia o edemas.
- El trasplante renal es el tratamiento de elección en la ERCT causada por NTAD ya que la enfermedad no recurre en el injerto.
- En los pacientes con mutación en HNF1B y con diabetes puede considerarse el doble trasplante renopancreático.
Conclusiones y conceptos claveSe describen en la (Tabla 5)






{kind=link}
{kind=link}
{kind=link}