
This is a case report of a 73-year-old man with new-onset acute renal failure while being investigated for pulmonary infiltrates and mediastinal lymphadenopathies. Urine tests showed tubular range proteinuria with no microhaematuria. Immunology tests showed elevated serum IgG and hypocomplementaemia (classical pathway activation). Renal biopsy and clinical-pathological correlation were crucial in this case, reinforcing their important role in the final diagnosis of acute kidney injury.
El caso presentado es el de un paciente varón de 73 años que debuta con un fracaso renal agudo en el contexto de infiltrados pulmonares y adenopatías mediastínicas a estudio. En el análisis de orina destacó proteinuria de rango tubular, sin microhematuria. En el estudio inmunológico se observó únicamente una elevación de los valores normales de IgG, junto con una activación de la vía clásica del complemento. La biopsia renal y la correcta correlación clínico-patológica fueron definitivas en este caso, mostrando una vez más ser una herramienta fundamental en el diagnóstico del fracaso renal agudo de etiología no clara.
A 73-year-old male patient was referred to our Nephrology department with acute renal failure in the context of constitutional syndrome. His serum creatinine level was 4.9mg/dL at the time of referral.
The patient had a 10-year history of hypertension, which was well controlled with losartan/ hydrochlorothiazide (100mg/25mg per day). He was also taking tamsulosin (0.4mg per day) for benign prostatic hypertrophy, which was monitored by his urologist.
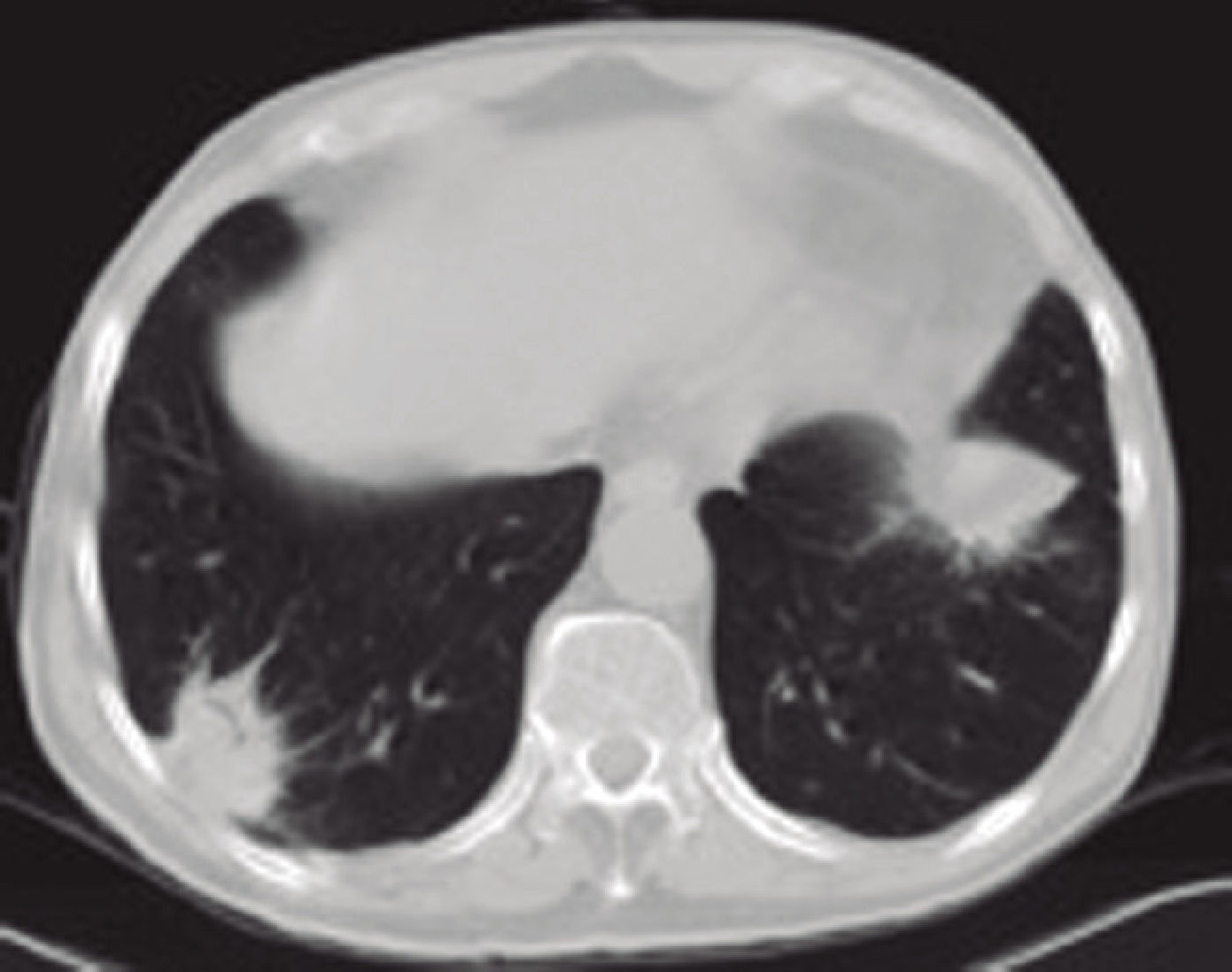
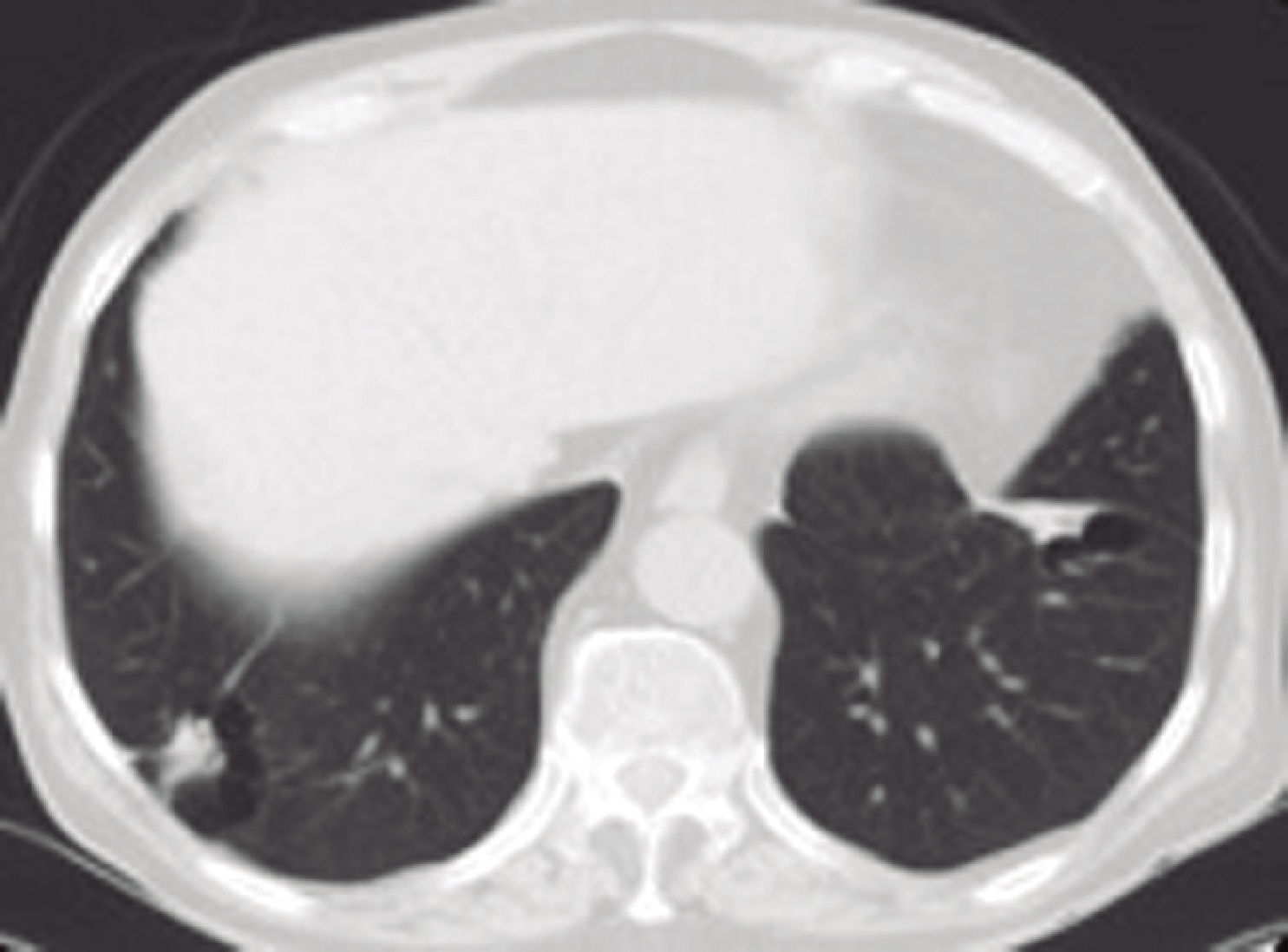
For the last 8 months, the patient had had a dry cough, accompanied by asthenia, anorexia and a weight loss of 6 kilograms. In view of the constitutional syndrome with cough, outpatient investigations had been undertaken, showing an initial creatinine level of 3.5mg/dL, which increased to 4.9mg/dL 10 days later. Renal ultrasound revealed normalsized kidneys with correct corticomedullary differentiation, and a thoracic CT scan showed lesions compatible with two pulmonary masses. One was located in the anterobasal segment of the left lower lobe (LLL) and measured approximately 5 × 4cm. The other was located in the posterobasal segment of the right lower lobe (RLL), measured approximately 4 × 3cm, had poorly-defined edges, and an air bronchogram in its interior. There was also slight peripheral infiltrate in the apical region of the RLL, with a non-specific inflammatory appearance. Another finding of note was the presence of mediastinal lymphadenopathies. A bronchoscopy was then performed, showing moderate infiltration of the subsegmental spur of the right basal segment, which was biopsied. Bronchial aspirate samples were taken for microbiology and cytology. Ziehl-Neelsen staining was negative. An attempt to perform a fine-needle aspiration biopsy of one of the pulmonary masses was unsuccessful due to technical problems; therefore, an in-patient mediastinoscopy was arranged with the Thoracic Surgery Department.
Finally, the patient was admitted to the Nephrology Department for study, diagnosis and treatment of acute renal failure, with progressive deterioration of renal function associated with pulmonary and lymph node involvement of unknown origin. On admission, the patient's blood pressure was normal and no enlarged cervical, axillary or femoral lymph nodes were palpated. The only observation of note was the patient's emaciated appearance.
Further investigationsBlood test results showed pancytopenia, leucocytes 3,100 × 109/L, haemoglobin 10.1g/dL, haematocrit 28.9%, and platelets 117,000 × 109/L. The patient's renal function had deteriorated, with serum creatinine reaching 6.64mg/dL (MDRD-eGFR 8ml/ min). The urine protein-to-creatinine ratio was 638mg/g and there was no evidence of microhaematuria. The immunology study showed an increased IgG and complement utilisation through classical pathway activation: IgG = 3,770 (700-1,400), C3/C4 = 33mg/dL (90-180)/<1mg/dL (10-40). A subsequent analysis of IgG subclasses revealed that the increase was mainly in IgG4: IgG1/IgG2/ IgG3/IgG4 = 1,813 (490-890)/557 (189-527)/156 (20-72)/1,244 (18-85) mg/dL.
The protein analysis showed beta-gamma bridging. The bone marrow aspirate ruled out monoclonal gammopathies. We repeated the thoracic CT scan and confirmed the previously described pulmonary nodular images and mediastinal lymphadenopathies. The thoracic surgeons performed a mediastinoscopy with lymph node biopsy; lymphomatous disease was ruled out in the histology report.
Lastly, in view of the unknown origin of the patient's progressive renal failure, a renal biopsy was taken, and this led to the final diagnosis.
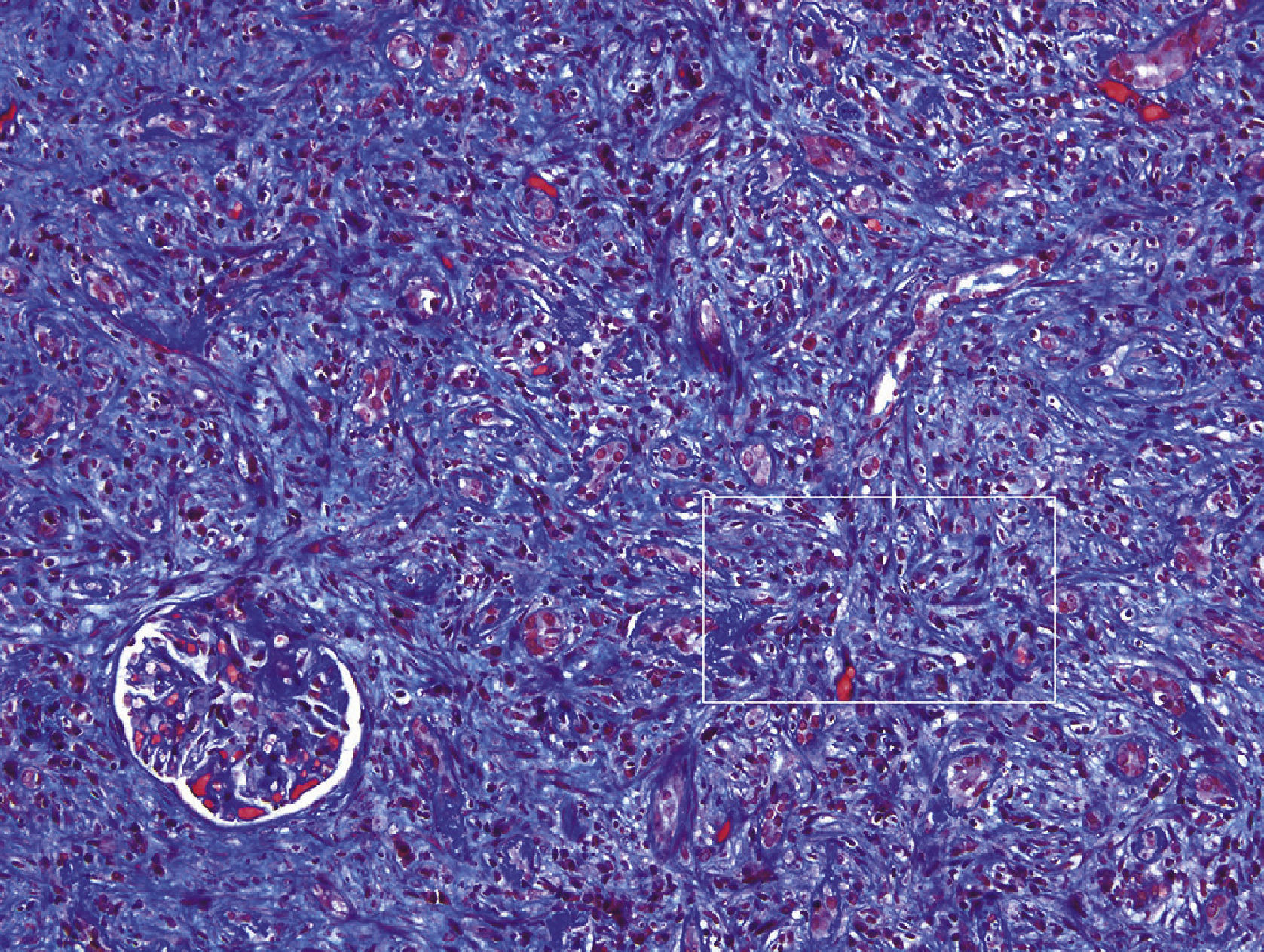
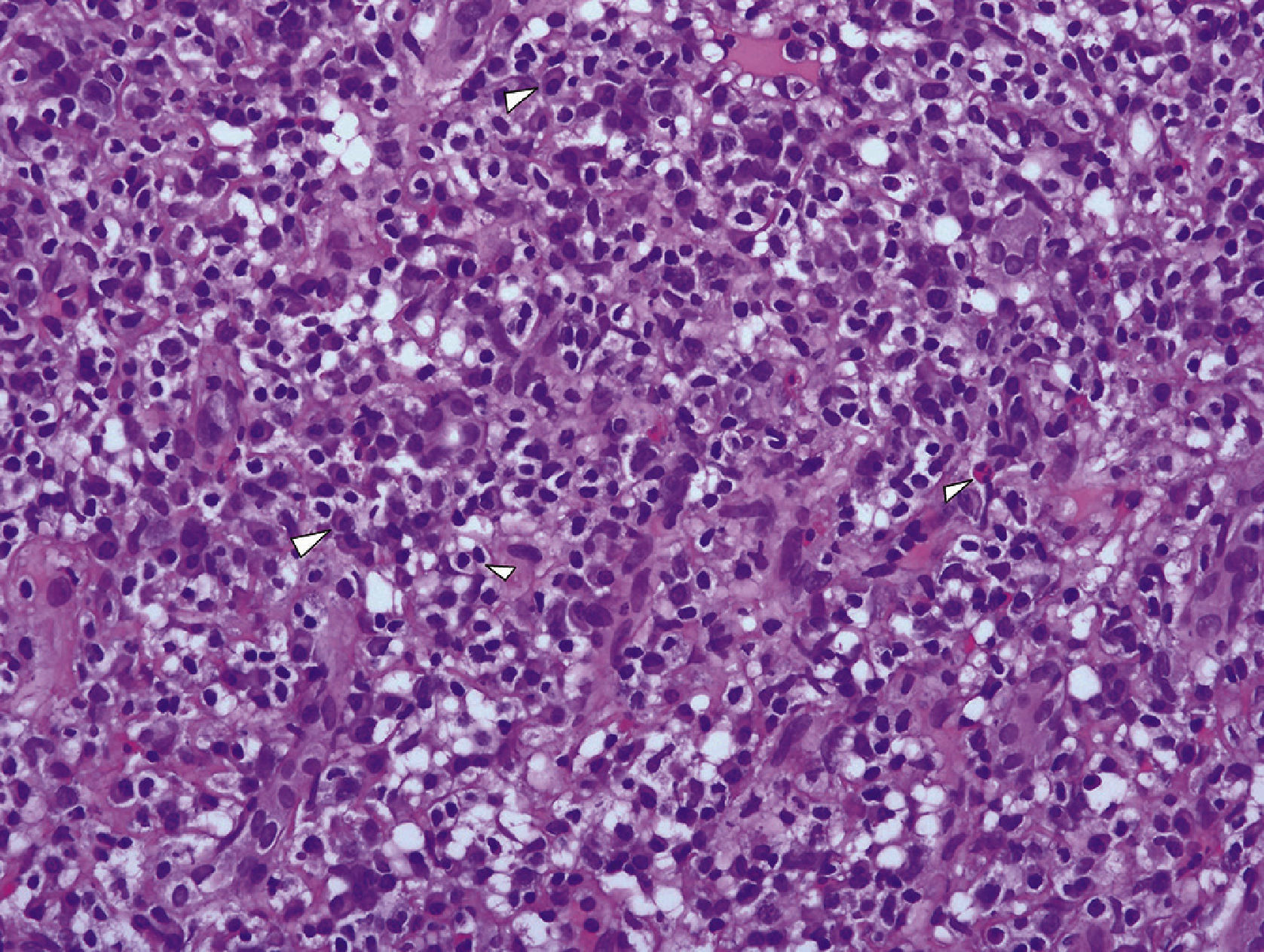
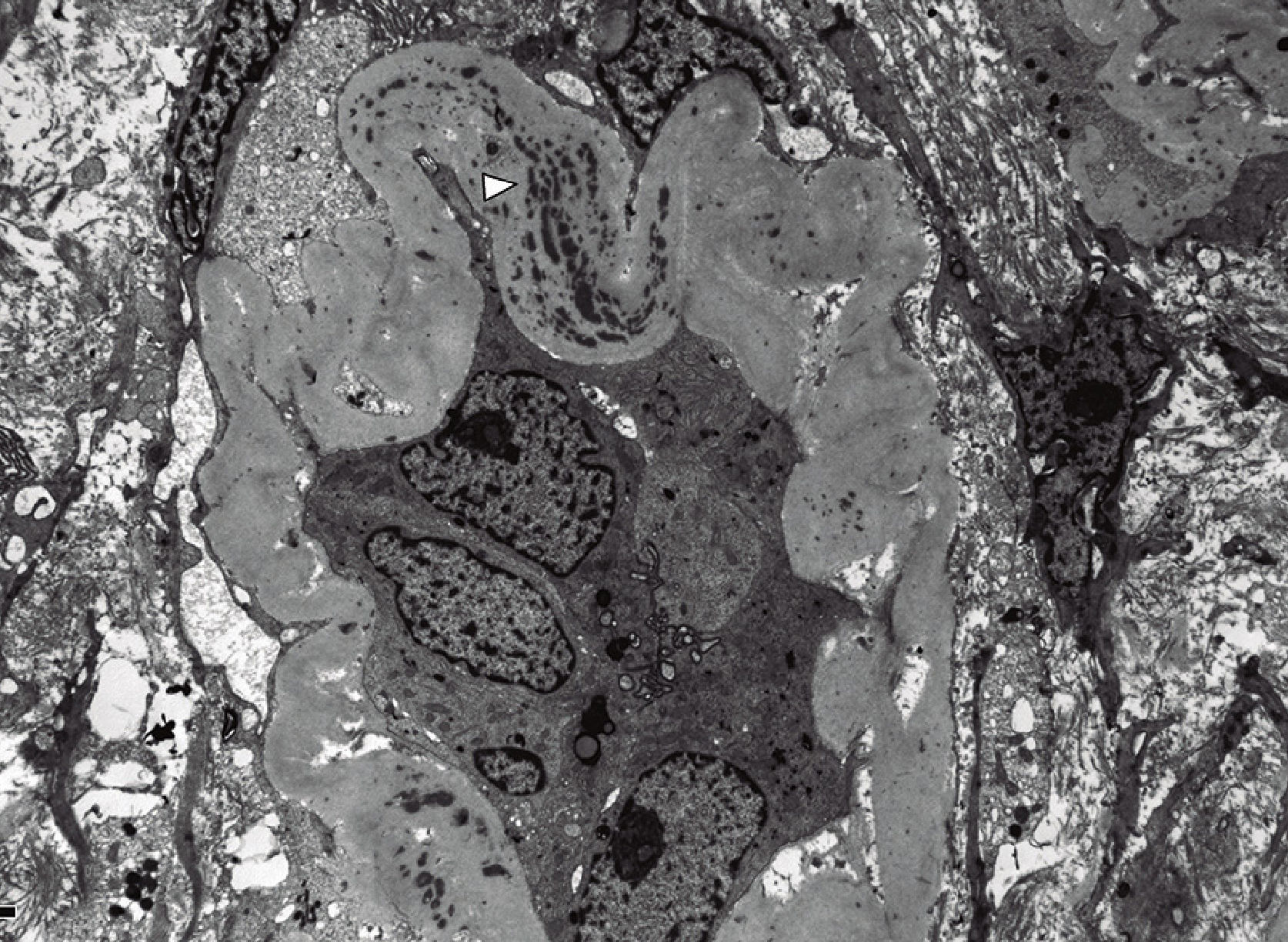
Anatomic pathology study of the renal biopsyDense, diffuse inflammatory infiltrate was observed in the cortical and medullary interstitial space, with abundant plasma cells and lymphocytes, and some eosinophils, as well as diffuse interstitial fibrosis (affecting over 50% of the cortical tissue) and tubular atrophy. The fibrosis had a focal storiform pattern - whereby the fibroblasts are arranged in bundles resembling the whorls of a straw mat - surrounding cell nests of lymphocytes and/or plasma cells.
Of the 15 glomeruli present in the sample, 9 were sclerosed and the rest had normal cells, with a slight mesangial matrix expansion. Some thick, folded basement membranes were observed segmentally. The interlobular arteries presented a narrowed lumen caused by marked fibrosis of their inner layer.
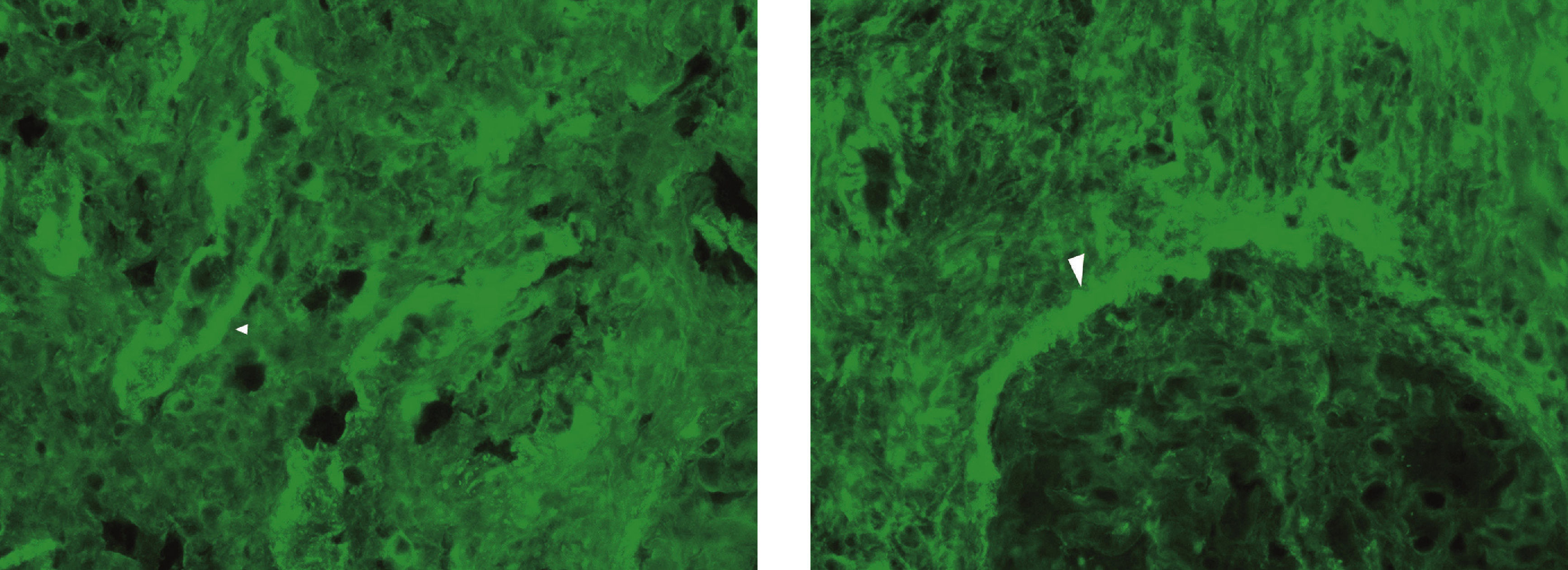
Immunofluorescence assay showed granular positivity for IgG, kappa, lambda and C3 (all with intensity 3+) in the tubular basement membranes and in the Bowman capsule. The 3 glomeruli in the sample were negative for IgG, IgA, IgM, C3, C1q, kappa and lambda.
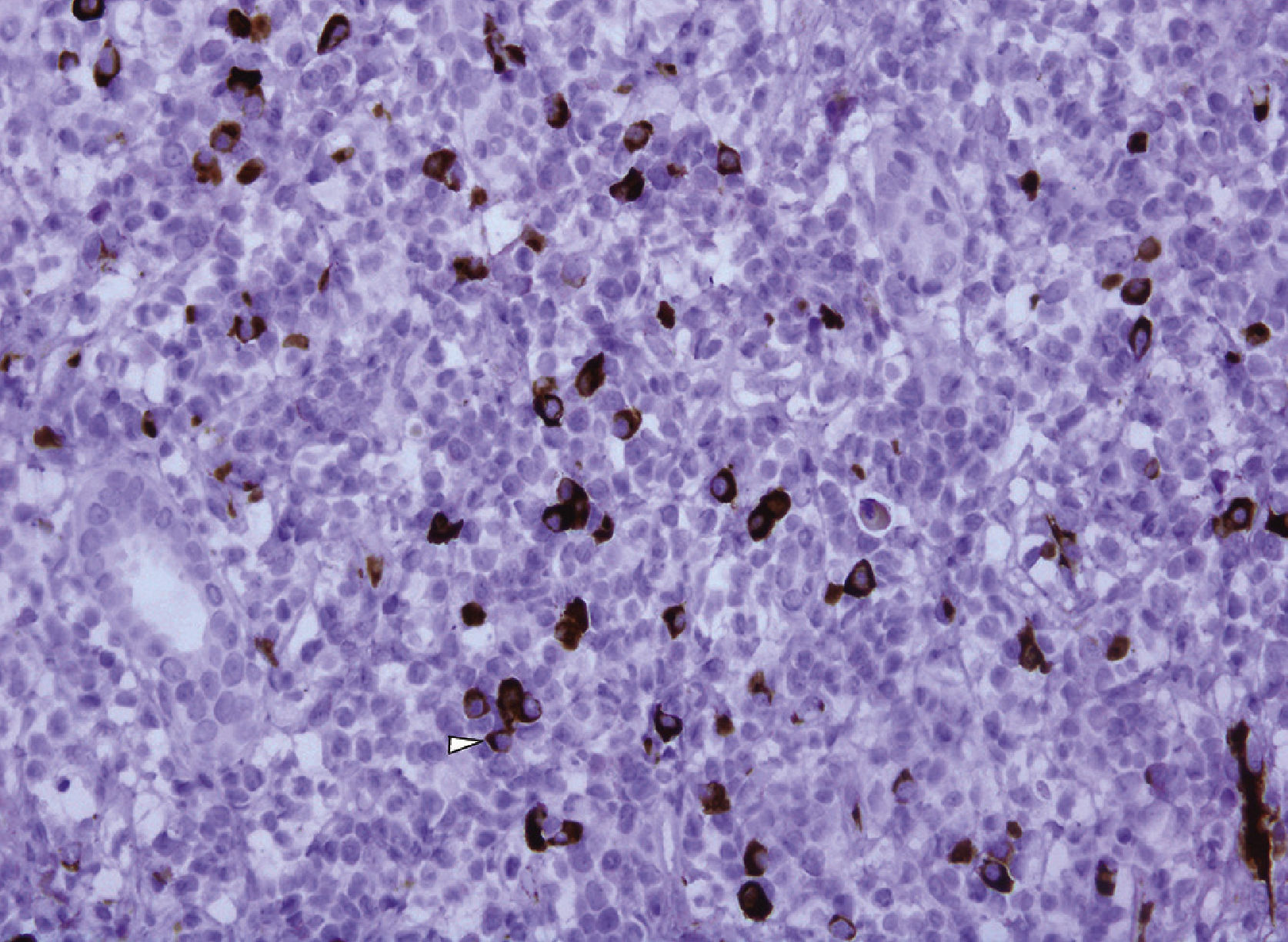
The immunohistochemical IgG4 study showed 10 to 20 IgG4+ plasma cells per high power field. The lymph cell infiltrate was predominantly T type.
The ultrastructural examination identified abundant electron-dense and amorphous deposits in the interior of thickened tubular basement membranes. The only glomerulus in the ultrastructural study sample had thick, folded basement membranes, with a slight fusion of the podocytes. No electron-dense deposits were observed.
Anatomic pathology study of the mediastinal lymph node biopsiesThe lymph nodes showed paracortical and follicular hyperplasia changes, as well as abundant plasma cell infiltrate.
The immunohistochemical analysis confirmed that the plasma cells were polyclonal; kappa and lambda light chains were positive. Staining showed more than 100 IgG4+ plasma cells per high power field.
OutcomeThe patient was treated with corticoids at a dose of 0.6mg/ kg, which was later tapered down to 5mg per day. Despite this, the patient is currently receiving haemodialysis, but has a good quality of life, as he is able to carry out his activities of daily living completely independently. It is likely that the extensive, chronic interstitial lesions seen in the biopsy contributed to the lack of renal recovery. Plasma IgG4 decreased to 1,074mg/dL at the beginning of treatment, and was 159mg/ dL 7 months later. A follow-up thoracic CT scan was performed after 8 months of the corticoid treatment, showing a reduction of the nodular lesions and no enlarged lymph nodes.
DiscussionOur patient was referred to the Nephrology Department due to acute renal failure, while being investigated for pulmonary masses and mediastinal lymphadenopathies. The clinical-pathological correlation of this case led to the diagnosis of IgG4 nephropathy.
IgG4-related disease (IgG4-RD) is a systemic autoimmune process that has only recently been recognised. It usually presents as inflammatory masses in multiple locations. It has a typical histological appearance, which is similar in all affected organs, characterised by a lymphoplasmacytic infiltrate rich in IgG4+ plasma cells, storiform fibrosis, and often, but not always, elevated serum IgG4.1,2 IgG4-RD was initially described in the pancreas, but it can affect any organ. The clinical manifestations vary depending on disease severity and organs affected.
IgG4-RD is more prevalent among males than females, with a ratio of 3:1, and it is more frequent in patients over the age of 50.
In the kidney, it can present as nodules or as diffuse infiltration in enlarged kidneys.3 The diagnosis is reached by renal biopsy, which shows tubulointerstitial nephritis (TIN) rich in plasma cells expressing IgG4, with abundant eosinophils in some cases. The presence of deposits in the tubular basement membranes is also a diagnostic criterion for this disease.4 As TIN is non-specific, it is easy to underdiagnose IgG4-RD unless an adequate clinical-pathological correlation is established.5
In the largest series of patients with IgG4-RD (n=153), Saeki et al reported that approximately 20% of cases had renal involvement confirmed by biopsy,6 which often revealed an IgG4+ plasma cell-rich lymphoplasmatic infiltrate. Moreover, up to 70% of the patients in this series had elevated serum IgG4 levels, hypergammaglobulinaemia and hypocomplementaemia.7 Furthermore, some of these patients did not have autoimmune pancreatitis.
The IgG4 molecule only accounts for 5% of the total concentration of IgG under normal conditions. IgG4 has a wide range of plasma concentrations, which range between 0.01 and 1.40mg/dL. In contrast to those of other IgG subclasses, the heavy chains (Fc region) of IgG 4 are bound by weak disulphide bridges. This facilitates IgG4 division and the formation of “hybrid” IgG4 molecules with 2 Fc and 2 light chains (Fab) with an affinity for different antigens. This reduces their propensity to form immune complexes, but they show a notable tendency to mimic rheumatoid factor activity, binding to each other through the Fc. IgG4 is produced when there is repeated and/or prolonged exposure to certain antigens (in infections and allergies). IgG4 production is controlled by the T helper 2 cell response.1 From an immunological standpoint, our patient initially had very elevated IgG4 levels. In addition, we observed an activation of the classical complement pathway (low C3 and C4), in common with other cases, and which seems to be related to the presence of circulating immune complexes. The literature describes IgG4-related disease as an autoimmune pathology triggered by a reaction against certain autoantigens (such as lactoferrin and carbonic anhydrase II). The autoantibodies formed in the inflamed organs could bind to epithelial antigens, leading to immune complex deposits on the epithelial basement membranes. Furthermore, although IgG4 does not activate the classical complement pathway by itself, it seems that other isoforms present in the kidney (such as IgG1 and IgG3) could activate it. Also, it appears that IgG4 could activate the complement cascade through the lecithin pathway.8
In addition to its role in IgG4-RD, IgG4 has also been associated with other diseases, such as Mikulicz syndrome, Riedel's thyroiditis, Ormond's disease, retroperitoneal fibrosis and inflammatory pseudotumours. In the kidney, IgG4-RD is manifested as TIN, but there have also been cases of renal involvement in the form of membranous glomerulonephritis (GN) (9%), IgA GN and nonspecific GN.6,9
Clinically, Ig G 4 -RD manifests as constitutional syndrome, with fever and elevated acute phase reactants, as well as chance findings in radiographic images. In cases involving the kidneys, patients have proteinuria in a nonnephrotic range with or without renal failure. Some patients have allergic symptoms (asthma, eczema, atopy, eosinophilia) and up to 40% of cases also present sinusitis and bronchial asthma. Our patient had primary involvement of the lungs, kidneys and mediastinal lymph nodes, and his rapidly progressive renal failure was particularly striking in this context.
Given the rarity of the disease, there is no scientific evidence regarding treatment, but usually the recommended therapy is based on corticosteroids.10 In general, patients are treated when the lymphoplasmacytic infiltrates cause target organ dysfunction (kidney dysfunction, in our case, led to renal failure). Caution should be exercised when treating asymptomatic patients and those with only enlarged lymph nodes, since the drugs involved have well-known adverse effects. Glucocorticoids are administered at doses of 0.6mg/ kg, and the dose is reduced progressively after 2 to 4 weeks, leaving the patient with a prednisone dose of 2.5-5mg/day for 3 years. Azathioprine, mycophenolate mofetil and methotrexate have been used as maintenance therapy with no clear scientific evidence. There have also been some cases of resistant disease, as well as recurrences, which have been treated with rituximab.11
The key determinant of treatment response is usually the level of fibrosis of the affected organ. IgG4 plasma levels do not return to normal values in 63% of cases, of which only 30% present recurrences. In some cases, monitoring of IgG4 levels could be used to identify recurrences.
Conflicts of InterestThe authors have no conflicts of interest to declare.
.")
.")
.")
.")
.")





